celltalker example
Last updated: 2020-11-24
Checks: 7 0
Knit directory: interaction-tools/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20191213) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version c6b67b8. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: .drake/
Ignored: data/COMUNET/
Ignored: data/CellChat/
Ignored: data/ICELLNET/
Ignored: data/NicheNet/
Ignored: data/cellphonedb/
Ignored: data/celltalker/
Ignored: output/14-CellChat.Rmd/
Ignored: output/15-talklr.Rmd/
Ignored: output/16-CiteFuse.Rmd/
Ignored: output/17-scTHI.Rmd/
Ignored: output/18-celltalker.Rmd/
Ignored: output/index.Rmd/
Ignored: renv/library/
Ignored: renv/python/
Ignored: renv/staging/
Untracked files:
Untracked: analysis/_navbar.html
Untracked: code/render.R
Unstaged changes:
Modified: _drake.R
Modified: analysis/_site.yml
Modified: analysis/index.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/18-celltalker.Rmd) and HTML (docs/18-celltalker.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | b305bfe | Luke Zappia | 2020-11-10 | Add celltalker to drake |
| Rmd | 4e1d47f | Luke Zappia | 2020-11-10 | Add celltalker tutorial |
# Setup document
source(here::here("code", "setup.R"))
# Function dependencies
invisible(drake::readd(download_link))Introduction
In this document we are going to run through the example analysis for the celltalker package and have a look at the output it produces. More information about celltalker can be found at https://github.com/arc85/celltalker.
library("celltalker")Chunk time: 0.15 secs
1 Input
1.1 Expression
The main input to celltalker is an expression matrix for a set of samples (3 PBMC samples and 3 tonsil samples) from GEO accession GSE139324. We create this by reading in the raw Cell Ranger output files and storing them as a Seurat object.
Note: It is not entirely clear from the celltalker tutorial exactly which samples to use so these results may differ from what is show there.
data_paths <- fs::dir_ls(here("data", "celltalker"))
names(data_paths) <- c("pbmc1", "pbmc2", "pbmc3", "tonsil1", "tonsil2",
"tonsil3")
raw_data <- Seurat::Read10X(data_paths)
metadata <- data.frame(
Sample = stringr::str_remove(colnames(raw_data), "_[A-Z-0-9]+")
)
metadata$Tissue <- stringr::str_remove(metadata$Sample, "[0-9]")
rownames(metadata) <- colnames(raw_data)
seurat <- Seurat::CreateSeuratObject(counts = raw_data, meta.data = metadata)
seuratAn object of class Seurat
33694 features across 15524 samples within 1 assay
Active assay: RNA (33694 features, 0 variable features)Chunk time: 15.36 secs
1.2 Ligand-receptor pairs
celltalker includes a set of ligand-receptor pairs.
skim(ramilowski_pairs)| Name | ramilowski_pairs |
| Number of rows | 2557 |
| Number of columns | 3 |
| _______________________ | |
| Column type frequency: | |
| factor | 3 |
| ________________________ | |
| Group variables | None |
Variable type: factor
| skim_variable | n_missing | complete_rate | ordered | n_unique | top_counts |
|---|---|---|---|---|---|
| ligand | 0 | 1 | FALSE | 708 | CAL: 41, GNA: 38, FN1: 28, CAL: 22 |
| receptor | 0 | 1 | FALSE | 691 | ITG: 67, ITG: 35, EGF: 32, LRP: 31 |
| pair | 0 | 1 | FALSE | 2557 | A2M: 1, AAN: 1, AAN: 1, ACE: 1 |
Chunk time: 0.09 secs
2 Seurat analysis
We first need to perform a standard Seurat analysis to get clusters and cell type labels. Most of the details are skipped over here.
2.1 Clustering
set.seed(02221989)
seurat <- Seurat::NormalizeData(seurat)
seurat <- Seurat::FindVariableFeatures(seurat)
seurat <- Seurat::ScaleData(seurat)
seurat <- Seurat::RunPCA(seurat, verbose = FALSE)
seurat <- Seurat::RunUMAP(seurat, reduction = "pca", dims = 1:15,
verbose = FALSE)
seurat <- Seurat::FindNeighbors(seurat, reduction = "pca", dims = 1:15,
verbose = FALSE)
seurat <- Seurat::FindClusters(seurat, resolution = 0.5, verbose = FALSE)
p1 <- Seurat::DimPlot(seurat, reduction = "umap", group.by = "Sample")
p2 <- Seurat::DimPlot(seurat, reduction = "umap", group.by = "Tissue")
p3 <- Seurat::DimPlot(seurat, reduction = "umap", group.by = "RNA_snn_res.0.5",
label = TRUE) +
Seurat::NoLegend()
cowplot::plot_grid(p1, p2, p3)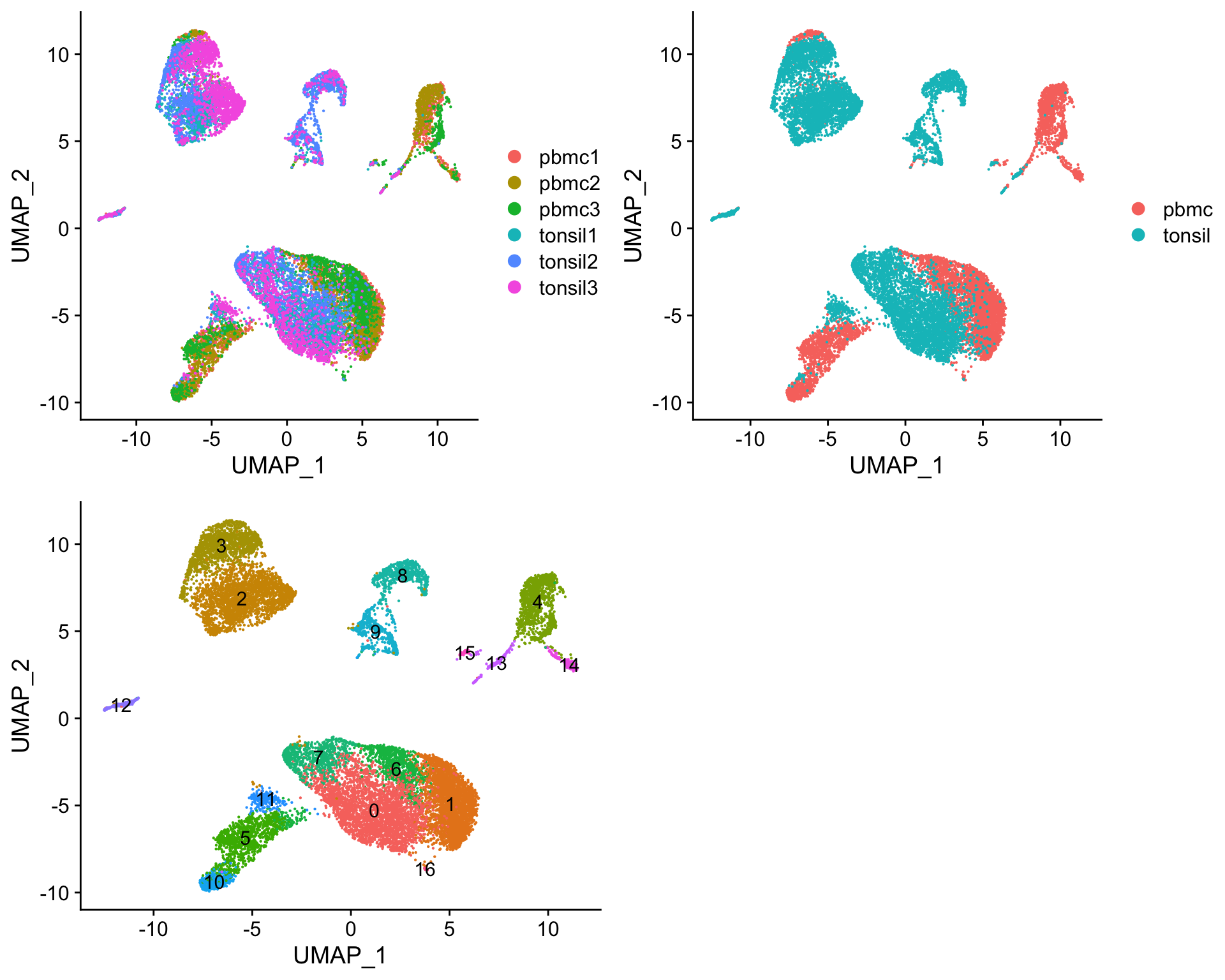
| Version | Author | Date |
|---|---|---|
| b305bfe | Luke Zappia | 2020-11-10 |
Chunk time: 52.39 secs
2.2 Labels
Seurat::FeaturePlot(seurat, reduction = "umap",
features = c("CD3D", "CD8A", "CD4", "CD14", "MS4A1",
"FCGR3A", "IL3RA"))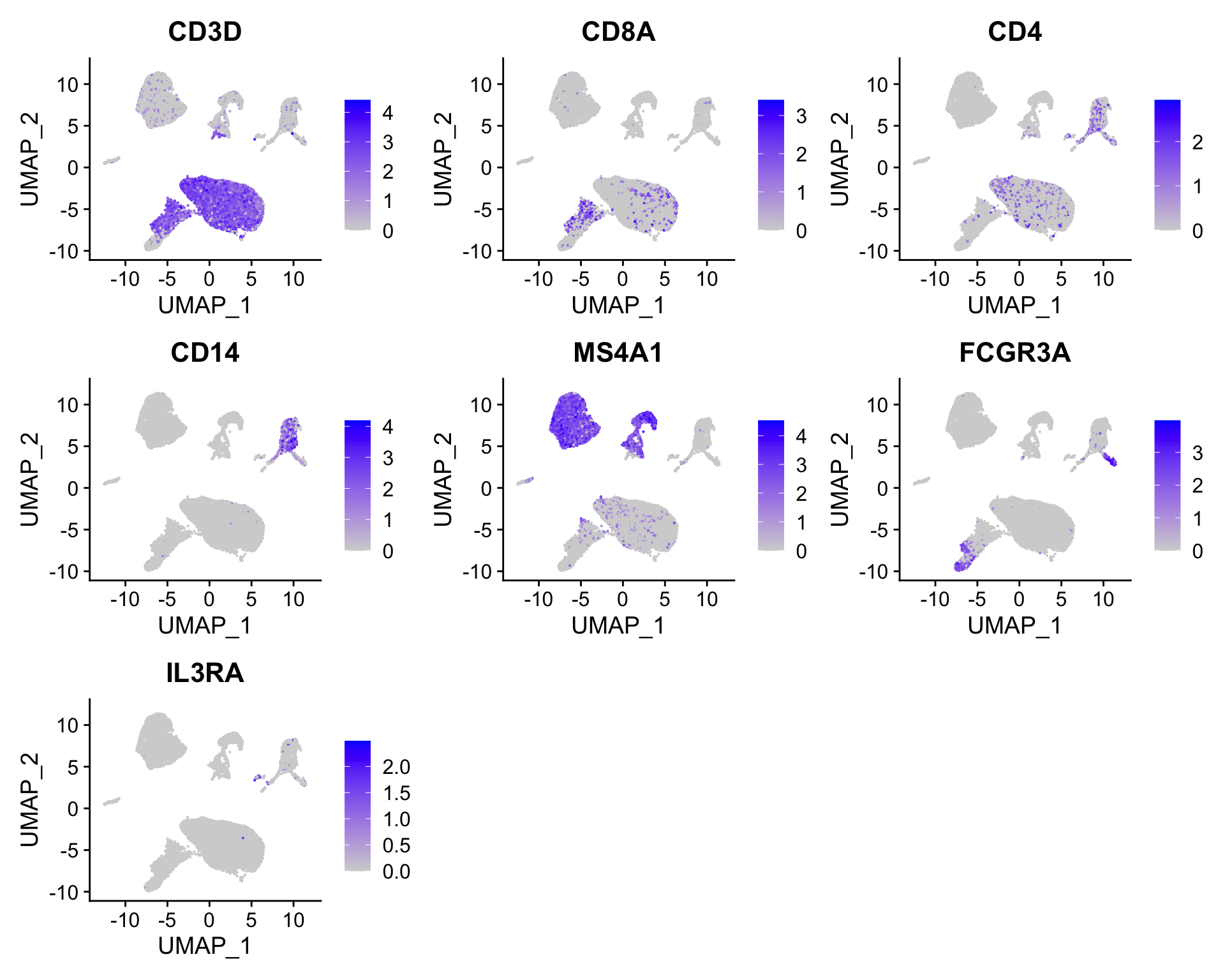
| Version | Author | Date |
|---|---|---|
| b305bfe | Luke Zappia | 2020-11-10 |
# Add metadata for cell types
cell_types <- vector("logical", length = ncol(seurat))
names(cell_types) <- colnames(seurat)
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "0"] <- "CD4 T conv"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "1"] <- "CD4 T conv"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "2"] <- "B cells"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "3"] <- "B cells"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "4"] <- "CD14 monocytes"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "5"] <- "CD8"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "6"] <- "CD4 T conv"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "7"] <- "CD4 T conv"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "8"] <- "B cells"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "9"] <- "B cells"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "10"] <- "NK cells"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "11"] <- "CD8"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "12"] <- "Plasma"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "13"] <- "CD14 monocytes"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "14"] <- "CD16 monocytes"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "15"] <- "PDC"
cell_types[seurat@meta.data$RNA_snn_res.0.5 == "16"] <- "RBCs"
seurat[["CellType"]] <- cell_types
# Remove red blood cells
rbc_cells <- names(cell_types)[seurat@meta.data$RNA_snn_res.0.5 == "16"]
seurat <- seurat [, !colnames(seurat) %in% rbc_cells]Chunk time: 8.66 secs
3 Select pairs
Before scoring interaction we first select a set of ligand-receptor pairs to look at. First we check which genes are present in the dataset and then we select those were the ligand or receptor is an up-regulated marker gene for one of the tissue types.
ligands <- as.character(unique(ramilowski_pairs$ligand))
receptors <- as.character(unique(ramilowski_pairs$receptor))
ligands_present <- rownames(seurat)[rownames(seurat) %in% ligands]
receptors_present <- rownames(seurat)[rownames(seurat) %in% receptors]
lr_present <- union(ligands_present, receptors_present)
Seurat::Idents(seurat) <- "Tissue"
markers <- Seurat::FindAllMarkers(seurat, assay="RNA", features = lr_present,
only.pos = TRUE, verbose = FALSE)
lr_sel <- unique(markers$gene)
interactions_sel <- rbind(
ramilowski_pairs[ramilowski_pairs$ligand %in% lr_sel, ],
ramilowski_pairs[ramilowski_pairs$receptor %in% lr_sel, ]
)Chunk time: 2.04 secs
This process selects 61 ligands and receptors and 241 interaction pairs.
4 Rearrange data
celltalker requires the input data in a specific structure. Helpfully it provides some useful functions to make this easier.
4.1 Expression matrices
The first data structure is a nested data frame with expression matrices for the selected ligands and receptors for each group, sample, and cluster.
expr_mat <- Seurat::GetAssayData(seurat, slot = "counts")
clusters <- seurat@meta.data$CellType
tissues <- seurat@meta.data$Tissue
samples <- seurat@meta.data$Sample
lr_matrices <- reshape_matrices(
count.matrix = expr_mat,
clusters = clusters,
groups = tissues,
replicates = samples,
ligands.and.receptors = interactions_sel
)
# There is a big where the next step fails if there are some clusters that
# aren't present in all groups. Add dummy values to avoid this.
dummy <- pull(unnest(lr_matrices, cols = "samples"))[[1]][[1]][0, ]
lr_matrices$samples[[2]]$expr.matrices <- map(
lr_matrices$samples[[2]]$expr.matrices, function(.exp_mats) {
missing <- unique(clusters[!(clusters %in% names(.exp_mats))])
for (cluster in missing) {
.exp_mats[[cluster]] <- dummy
}
.exp_mats
}
)
skim(lr_matrices)| Name | lr_matrices |
| Number of rows | 2 |
| Number of columns | 2 |
| _______________________ | |
| Column type frequency: | |
| character | 1 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| group | 0 | 1 | 4 | 6 | 0 | 2 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| samples | 0 | 1 | 2 | 2 | 2 |
skim(unnest(lr_matrices, cols = "samples"))| Name | unnest(lr_matrices, cols … |
| Number of rows | 6 |
| Number of columns | 3 |
| _______________________ | |
| Column type frequency: | |
| character | 2 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| group | 0 | 1 | 4 | 6 | 0 | 2 | 0 |
| sample | 0 | 1 | 5 | 7 | 0 | 6 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| expr.matrices | 0 | 1 | 6 | 8 | 8 |
Chunk time: 1.06 secs
4.2 Expressed ligands and receptors
From the nested expression matrices we then create a similar structure which contains the ligands and receptors that are expressed in each group/cluster combination.
lr_expressed <- create_lig_rec_tib(
exp.tib = lr_matrices,
clusters = clusters,
groups = tissues,
replicates = samples,
cells.reqd = 10,
freq.pos.reqd = 0.5,
ligands.and.receptors = interactions_sel
)
skim(lr_expressed)| Name | lr_expressed |
| Number of rows | 2 |
| Number of columns | 2 |
| _______________________ | |
| Column type frequency: | |
| character | 1 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| group | 0 | 1 | 4 | 6 | 0 | 2 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| lig.rec.exp | 0 | 1 | 2 | 2 | 2 |
skim(unnest(lr_expressed[1, 2], cols = "lig.rec.exp"))| Name | unnest(lr_expressed[1, 2]… |
| Number of rows | 8 |
| Number of columns | 2 |
| _______________________ | |
| Column type frequency: | |
| character | 1 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| cluster.id | 0 | 1 | 3 | 14 | 0 | 8 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| ligands.and.receptors | 0 | 1 | 8 | 2 | 2 |
Chunk time: 1.08 secs
5 Interactions
5.1 Putative interactions
With this data structure we can now look for putative interactions.
putative_interactions <- putative_interactions(
ligand.receptor.tibble = lr_expressed,
clusters = clusters,
groups = tissues,
freq.group.in.cluster = 0.05,
ligands.and.receptors = interactions_sel
)
skim(putative_interactions)| Name | putative_interactions |
| Number of rows | 2 |
| Number of columns | 2 |
| _______________________ | |
| Column type frequency: | |
| character | 1 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| group | 0 | 1 | 4 | 6 | 0 | 2 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| lig_rec_list | 0 | 1 | 2 | 302 | 304 |
Chunk time: 1.31 secs
The nested data frame returned by this function contains lists of the active ligands and receptors for each cell type in each group.
5.2 Unique interactions
The next step identifies interactions that are unique to each group.
unique_interactions <- unique_interactions(
putative_interactions,
group1 = "pbmc",
group2 = "tonsil",
interactions_sel
)
skim(unique_interactions)| Name | unique_interactions |
| Number of rows | 3 |
| Number of columns | 2 |
| _______________________ | |
| Column type frequency: | |
| character | 1 |
| list | 1 |
| ________________________ | |
| Group variables | None |
Variable type: character
| skim_variable | n_missing | complete_rate | min | max | empty | n_unique | whitespace |
|---|---|---|---|---|---|---|---|
| comparison | 0 | 1 | 6 | 9 | 0 | 3 | 0 |
Variable type: list
| skim_variable | n_missing | complete_rate | n_unique | min_length | max_length |
|---|---|---|---|---|---|
| ligands.and.receptors | 0 | 1 | 3 | 22 | 186 |
Chunk time: 0.04 secs
This gives use the ligands and receptors that are unique to each group as well as those they have in common.
6 Visualisation
6.1 Circos plot
6.1.1 PBMC
pbmc_lr <- pull(unique_interactions[1, 2])[[1]]
pbmc_interactions <- pull(putative_interactions[1, 2])[[1]][pbmc_lr]
circos_plot(interactions = pbmc_interactions, clusters = clusters)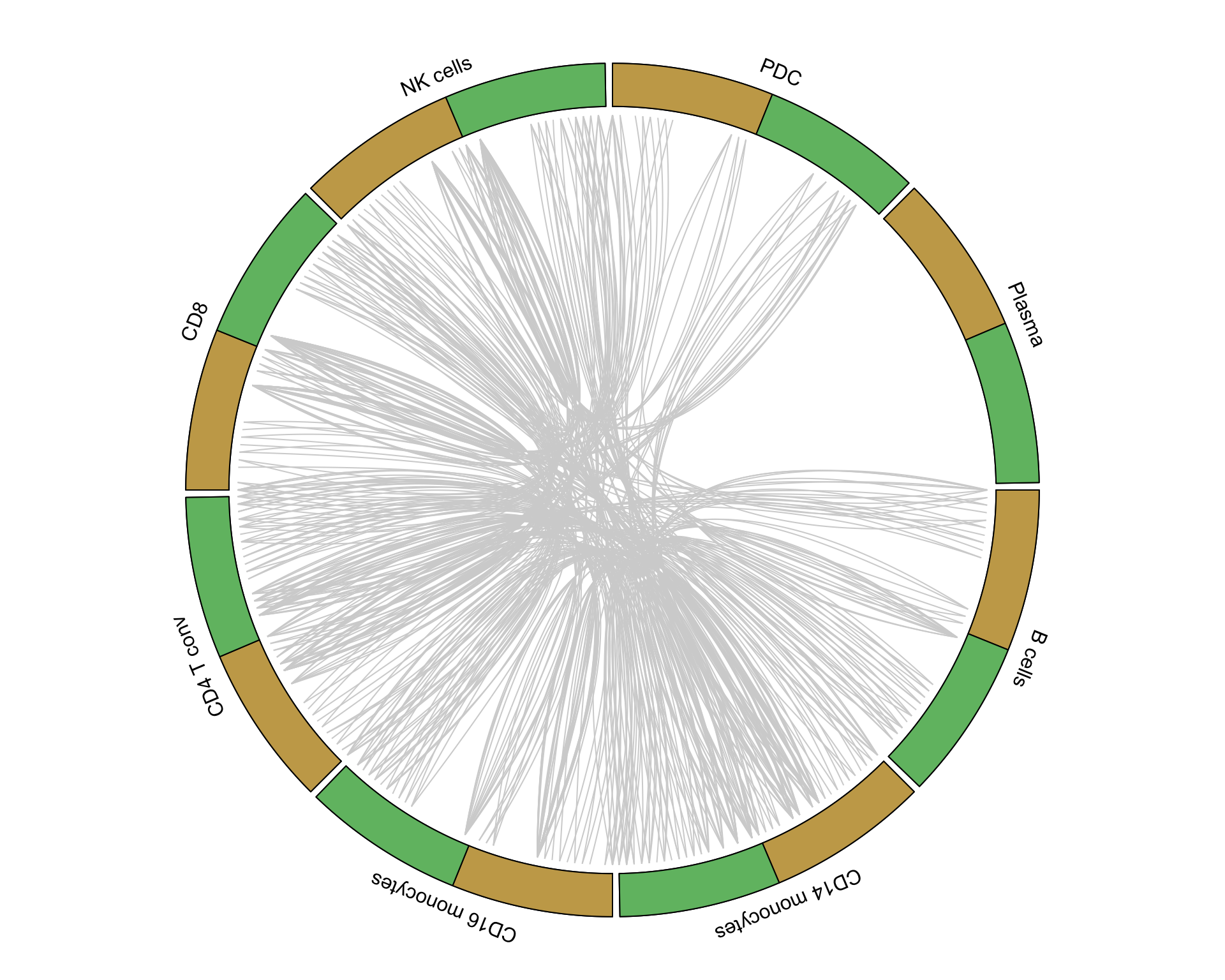
| Version | Author | Date |
|---|---|---|
| b305bfe | Luke Zappia | 2020-11-10 |
[1] "Plotted interactions"Chunk time: 1.49 secs
6.1.2 Tonsil
tonsil_lr <- pull(unique_interactions[2, 2])[[1]]
tonsil_interactions <- pull(putative_interactions[2, 2])[[1]][tonsil_lr]
circos_plot(interactions = tonsil_interactions, clusters = clusters)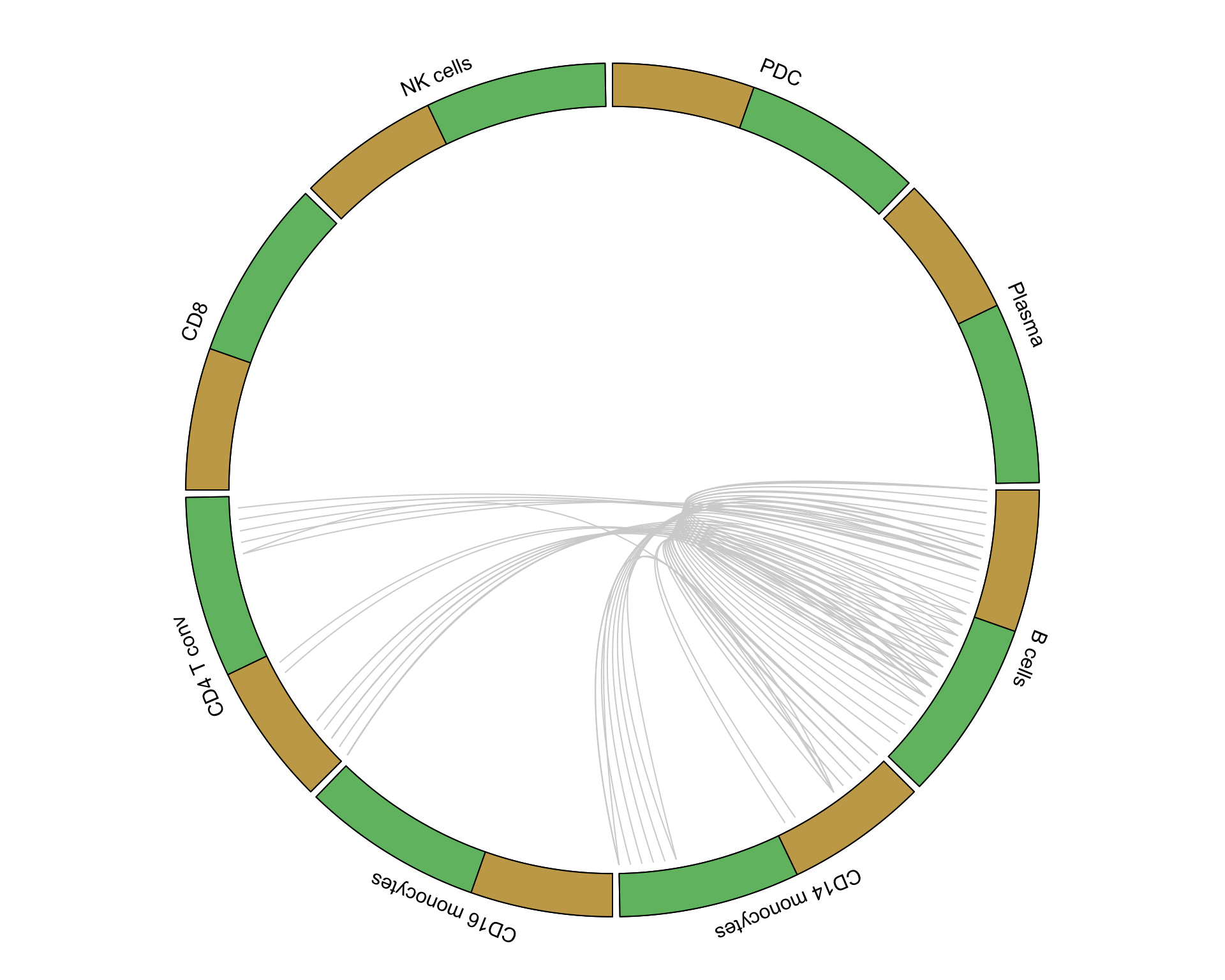
| Version | Author | Date |
|---|---|---|
| b305bfe | Luke Zappia | 2020-11-10 |
[1] "Plotted interactions"Chunk time: 0.46 secs
Summary
Parameters
This table describes parameters used and set in this document.
params <- list(
)
params <- toJSON(params, pretty = TRUE)
kable(fromJSON(params))Chunk time: 0.01 secs
Output files
This table describes the output files produced by this document. Right click and Save Link As… to download the results.
kable(data.frame(
File = c(
download_link("parameters.json", OUT_DIR)
),
Description = c(
"Parameters set and used in this analysis"
)
))| File | Description |
|---|---|
| parameters.json | Parameters set and used in this analysis |
Chunk time: 0.01 secs
Session information
sessioninfo::session_info()─ Session info ───────────────────────────────────────────────────────────────
setting value
version R version 4.0.0 (2020-04-24)
os macOS Catalina 10.15.7
system x86_64, darwin17.0
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Europe/Berlin
date 2020-11-24
─ Packages ───────────────────────────────────────────────────────────────────
! package * version date lib source
abind 1.4-5 2016-07-21 [1] CRAN (R 4.0.0)
P assertthat 0.2.1 2019-03-21 [?] CRAN (R 4.0.0)
P backports 1.1.6 2020-04-05 [?] CRAN (R 4.0.0)
P base64enc 0.1-3 2015-07-28 [?] CRAN (R 4.0.0)
P base64url 1.4 2018-05-14 [?] standard (@1.4)
P broom 0.5.6 2020-04-20 [?] CRAN (R 4.0.0)
P cellranger 1.1.0 2016-07-27 [?] standard (@1.1.0)
P celltalker * 0.0.3.9000 2020-11-10 [?] Github (arc85/celltalker@533b28e)
P circlize * 0.4.9 2020-04-30 [?] CRAN (R 4.0.0)
P cli 2.0.2 2020-02-28 [?] CRAN (R 4.0.0)
P cluster 2.1.0 2019-06-19 [?] CRAN (R 4.0.0)
P codetools 0.2-18 2020-11-04 [?] CRAN (R 4.0.2)
P colorspace 1.4-1 2019-03-18 [?] standard (@1.4-1)
P conflicted * 1.0.4 2019-06-21 [?] standard (@1.0.4)
P cowplot 1.0.0 2019-07-11 [?] CRAN (R 4.0.0)
P crayon 1.3.4 2017-09-16 [?] CRAN (R 4.0.0)
P data.table 1.12.8 2019-12-09 [?] CRAN (R 4.0.1)
P DBI 1.1.0 2019-12-15 [?] CRAN (R 4.0.0)
P dbplyr 1.4.3 2020-04-19 [?] CRAN (R 4.0.0)
deldir 0.1-28 2020-07-15 [1] CRAN (R 4.0.2)
P digest 0.6.25 2020-02-23 [?] CRAN (R 4.0.0)
P dplyr * 0.8.5 2020-03-07 [?] CRAN (R 4.0.0)
P drake 7.12.0 2020-03-25 [?] CRAN (R 4.0.0)
P ellipsis 0.3.0 2019-09-20 [?] CRAN (R 4.0.0)
P evaluate 0.14 2019-05-28 [?] standard (@0.14)
P fansi 0.4.1 2020-01-08 [?] CRAN (R 4.0.0)
P farver 2.0.3 2020-01-16 [?] CRAN (R 4.0.0)
fastmap 1.0.1 2019-10-08 [1] CRAN (R 4.0.2)
P filelock 1.0.2 2018-10-05 [?] CRAN (R 4.0.0)
P fitdistrplus 1.0-14 2019-01-23 [?] CRAN (R 4.0.0)
P forcats * 0.5.0 2020-03-01 [?] CRAN (R 4.0.0)
P fs * 1.4.1 2020-04-04 [?] CRAN (R 4.0.0)
P future 1.17.0 2020-04-18 [?] CRAN (R 4.0.1)
P future.apply 1.5.0 2020-04-17 [?] CRAN (R 4.0.0)
P generics 0.0.2 2018-11-29 [?] standard (@0.0.2)
P ggplot2 * 3.3.0 2020-03-05 [?] CRAN (R 4.0.0)
ggrepel 0.8.2 2020-03-08 [1] CRAN (R 4.0.0)
ggridges 0.5.2 2020-01-12 [1] CRAN (R 4.0.0)
P git2r 0.27.1 2020-05-03 [?] CRAN (R 4.0.0)
P GlobalOptions 0.1.1 2019-09-30 [?] CRAN (R 4.0.0)
globals 0.12.5 2019-12-07 [1] CRAN (R 4.0.0)
P glue * 1.4.0 2020-04-03 [?] CRAN (R 4.0.0)
goftest 1.2-2 2019-12-02 [1] CRAN (R 4.0.2)
gridExtra 2.3 2017-09-09 [1] CRAN (R 4.0.0)
P gtable 0.3.0 2019-03-25 [?] standard (@0.3.0)
P haven 2.2.0 2019-11-08 [?] standard (@2.2.0)
P here * 0.1 2017-05-28 [?] standard (@0.1)
P highr 0.8 2019-03-20 [?] standard (@0.8)
P hms 0.5.3 2020-01-08 [?] CRAN (R 4.0.0)
P htmltools 0.5.0 2020-06-16 [?] CRAN (R 4.0.2)
htmlwidgets 1.5.1 2019-10-08 [1] CRAN (R 4.0.0)
P httpuv 1.5.2 2019-09-11 [?] standard (@1.5.2)
P httr 1.4.1 2019-08-05 [?] standard (@1.4.1)
ica 1.0-2 2018-05-24 [1] CRAN (R 4.0.0)
P igraph 1.2.5 2020-03-19 [?] CRAN (R 4.0.0)
irlba 2.3.3 2019-02-05 [1] CRAN (R 4.0.0)
P jsonlite * 1.6.1 2020-02-02 [?] CRAN (R 4.0.0)
P KernSmooth 2.23-17 2020-04-26 [?] CRAN (R 4.0.0)
P knitr * 1.28 2020-02-06 [?] CRAN (R 4.0.0)
P labeling 0.3 2014-08-23 [?] standard (@0.3)
P later 1.0.0 2019-10-04 [?] standard (@1.0.0)
P lattice 0.20-41 2020-04-02 [?] CRAN (R 4.0.0)
P lazyeval 0.2.2 2019-03-15 [?] standard (@0.2.2)
leiden 0.3.3 2020-02-04 [1] CRAN (R 4.0.0)
P lifecycle 0.2.0 2020-03-06 [?] CRAN (R 4.0.0)
P limma 3.44.1 2020-04-28 [?] Bioconductor
listenv 0.8.0 2019-12-05 [1] CRAN (R 4.0.0)
lmtest 0.9-37 2019-04-30 [1] CRAN (R 4.0.0)
lsei 1.2-0.1 2020-05-06 [1] CRAN (R 4.0.0)
P lubridate 1.7.8 2020-04-06 [?] CRAN (R 4.0.0)
P magrittr 1.5 2014-11-22 [?] CRAN (R 4.0.0)
P MASS 7.3-51.6 2020-04-26 [?] CRAN (R 4.0.0)
P Matrix 1.2-18 2019-11-27 [?] standard (@1.2-18)
matrixStats 0.56.0 2020-03-13 [1] CRAN (R 4.0.2)
P memoise 1.1.0 2017-04-21 [?] standard (@1.1.0)
P mgcv 1.8-31 2019-11-09 [?] standard (@1.8-31)
P mime 0.9 2020-02-04 [?] CRAN (R 4.0.0)
miniUI 0.1.1.1 2018-05-18 [1] CRAN (R 4.0.2)
P modelr 0.1.7 2020-04-30 [?] CRAN (R 4.0.0)
P munsell 0.5.0 2018-06-12 [?] standard (@0.5.0)
P nlme 3.1-147 2020-04-13 [?] CRAN (R 4.0.0)
npsurv 0.4-0.1 2020-05-06 [1] CRAN (R 4.0.0)
P pander * 0.6.3 2018-11-06 [?] CRAN (R 4.0.0)
P patchwork 1.0.0 2019-12-01 [?] CRAN (R 4.0.0)
pbapply 1.4-2 2019-08-31 [1] CRAN (R 4.0.0)
P pillar 1.4.4 2020-05-05 [?] CRAN (R 4.0.0)
P pkgconfig 2.0.3 2019-09-22 [?] CRAN (R 4.0.0)
plotly 4.9.2.1 2020-04-04 [1] CRAN (R 4.0.0)
P plyr 1.8.6 2020-03-03 [?] CRAN (R 4.0.0)
png 0.1-7 2013-12-03 [1] CRAN (R 4.0.0)
P polyclip 1.10-0 2019-03-14 [?] CRAN (R 4.0.0)
P prettyunits 1.1.1 2020-01-24 [?] CRAN (R 4.0.0)
P progress 1.2.2 2019-05-16 [?] CRAN (R 4.0.0)
P promises 1.1.0 2019-10-04 [?] standard (@1.1.0)
P purrr * 0.3.4 2020-04-17 [?] CRAN (R 4.0.0)
P R6 2.4.1 2019-11-12 [?] CRAN (R 4.0.0)
RANN 2.6.1 2019-01-08 [1] CRAN (R 4.0.0)
P RColorBrewer 1.1-2 2014-12-07 [?] standard (@1.1-2)
P Rcpp 1.0.4.6 2020-04-09 [?] CRAN (R 4.0.0)
RcppAnnoy 0.0.16 2020-03-08 [1] CRAN (R 4.0.0)
P readr * 1.3.1 2018-12-21 [?] standard (@1.3.1)
P readxl 1.3.1 2019-03-13 [?] standard (@1.3.1)
P renv 0.12.0 2020-08-28 [?] CRAN (R 4.0.2)
P repr 1.1.0 2020-01-28 [?] CRAN (R 4.0.0)
P reprex 0.3.0 2019-05-16 [?] standard (@0.3.0)
P reshape2 1.4.4 2020-04-09 [?] CRAN (R 4.0.0)
P reticulate 1.16 2020-05-27 [?] CRAN (R 4.0.2)
P rlang 0.4.6 2020-05-02 [?] CRAN (R 4.0.0)
P rmarkdown 2.1 2020-01-20 [?] CRAN (R 4.0.0)
ROCR 1.0-11 2020-05-02 [1] CRAN (R 4.0.0)
P rpart 4.1-15 2019-04-12 [?] CRAN (R 4.0.0)
P rprojroot 1.3-2 2018-01-03 [?] CRAN (R 4.0.0)
RSpectra 0.16-0 2019-12-01 [1] CRAN (R 4.0.0)
P rstudioapi 0.11 2020-02-07 [?] CRAN (R 4.0.0)
rsvd 1.0.3 2020-02-17 [1] CRAN (R 4.0.0)
Rtsne 0.15 2018-11-10 [1] CRAN (R 4.0.0)
P rvest 0.3.5 2019-11-08 [?] standard (@0.3.5)
P scales 1.1.0 2019-11-18 [?] standard (@1.1.0)
P sctransform 0.3.1 2020-10-08 [?] CRAN (R 4.0.2)
P sessioninfo 1.1.1 2018-11-05 [?] CRAN (R 4.0.0)
P Seurat 3.2.2 2020-09-26 [?] CRAN (R 4.0.2)
P shape 1.4.4 2018-02-07 [?] CRAN (R 4.0.0)
P shiny 1.5.0 2020-06-23 [?] CRAN (R 4.0.2)
P skimr * 2.1.1 2020-04-16 [?] CRAN (R 4.0.0)
spatstat 1.64-1 2020-05-12 [1] CRAN (R 4.0.2)
spatstat.data 1.4-3 2020-01-26 [1] CRAN (R 4.0.2)
spatstat.utils 1.17-0 2020-02-07 [1] CRAN (R 4.0.2)
P storr 1.2.1 2018-10-18 [?] standard (@1.2.1)
P stringi 1.4.6 2020-02-17 [?] CRAN (R 4.0.0)
P stringr * 1.4.0 2019-02-10 [?] CRAN (R 4.0.0)
P survival 3.2-7 2020-09-28 [?] CRAN (R 4.0.2)
tensor 1.5 2012-05-05 [1] CRAN (R 4.0.2)
P tibble * 3.0.1 2020-04-20 [?] CRAN (R 4.0.0)
P tidyr * 1.0.3 2020-05-07 [?] CRAN (R 4.0.0)
P tidyselect 1.0.0 2020-01-27 [?] CRAN (R 4.0.0)
P tidyverse * 1.3.0 2019-11-21 [?] standard (@1.3.0)
P txtq 0.2.0 2019-10-15 [?] standard (@0.2.0)
uwot 0.1.8 2020-03-16 [1] CRAN (R 4.0.0)
P vctrs 0.2.4 2020-03-10 [?] CRAN (R 4.0.0)
P viridisLite 0.3.0 2018-02-01 [?] standard (@0.3.0)
P whisker 0.4 2019-08-28 [?] standard (@0.4)
P withr 2.2.0 2020-04-20 [?] CRAN (R 4.0.0)
P workflowr 1.6.2 2020-04-30 [?] CRAN (R 4.0.0)
P xfun 0.13 2020-04-13 [?] CRAN (R 4.0.0)
P xml2 1.3.2 2020-04-23 [?] CRAN (R 4.0.0)
xtable 1.8-4 2019-04-21 [1] CRAN (R 4.0.2)
P yaml 2.2.1 2020-02-01 [?] CRAN (R 4.0.0)
zoo 1.8-8 2020-05-02 [1] CRAN (R 4.0.0)
[1] /Users/luke.zappia/Documents/Projects/interaction-tools/renv/library/R-4.0/x86_64-apple-darwin17.0
[2] /private/var/folders/rj/60lhr791617422kqvh0r4vy40000gn/T/RtmpOqMPdA/renv-system-library
[3] /private/var/folders/rj/60lhr791617422kqvh0r4vy40000gn/T/RtmpGqapVJ/renv-system-library
[4] /private/var/folders/rj/60lhr791617422kqvh0r4vy40000gn/T/RtmpgtsH80/renv-system-library
P ── Loaded and on-disk path mismatch.Chunk time: 0.32 secs