Convert between AnnData and SingleCellExperiment
Source:R/AnnData2SCE.R, R/SCE2AnnData.R
AnnData-Conversion.RdConversion between Python AnnData objects and SingleCellExperiment::SingleCellExperiment objects.
AnnData2SCE(
adata,
X_name = NULL,
layers = TRUE,
uns = TRUE,
var = TRUE,
obs = TRUE,
varm = TRUE,
obsm = TRUE,
varp = TRUE,
obsp = TRUE,
raw = FALSE,
skip_assays = FALSE,
hdf5_backed = TRUE,
verbose = NULL
)
SCE2AnnData(
sce,
X_name = NULL,
assays = TRUE,
colData = TRUE,
rowData = TRUE,
varm = TRUE,
reducedDims = TRUE,
metadata = TRUE,
colPairs = TRUE,
rowPairs = TRUE,
skip_assays = FALSE,
verbose = NULL
)Arguments
- adata
A reticulate reference to a Python AnnData object.
- X_name
For
SCE2AnnData()name of the assay to use as the primary matrix (X) of the AnnData object. IfNULL, the first assay ofscewill be used by default. ForAnnData2SCE()name used when savingXas an assay. IfNULLlooks for anX_namevalue inuns, otherwise uses"X".- layers, uns, var, obs, varm, obsm, varp, obsp, raw
Arguments specifying how these slots are converted. If
TRUEeverything in that slot is converted, ifFALSEnothing is converted and if a character vector only those items or columns are converted.- skip_assays
Logical scalar indicating whether to skip conversion of any assays in
sceoradata, replacing them with empty sparse matrices instead.- hdf5_backed
Logical scalar indicating whether HDF5-backed matrices in
adatashould be represented as HDF5Array objects. This assumes thatadatais created withbacked="r".- verbose
Logical scalar indicating whether to print progress messages. If
NULLusesgetOption("zellkonverter.verbose").- sce
- assays, colData, rowData, reducedDims, metadata, colPairs, rowPairs
Arguments specifying how these slots are converted. If
TRUEeverything in that slot is converted, ifFALSEnothing is converted and if a character vector only those items or columns are converted.
Value
AnnData2SCE() will return a
SingleCellExperiment::SingleCellExperiment containing the equivalent data
from adata.
SCE2AnnData() will return a reticulate reference to an AnnData object
containing the content of sce.
Details
Python environment
These functions assume that an appropriate Python environment has already been loaded. As such, they are largely intended for developer use, most typically inside a basilisk context.
Conversion mapping
The conversion is not entirely lossless. The current mapping is shown below (also at https://tinyurl.com/AnnData2SCE):
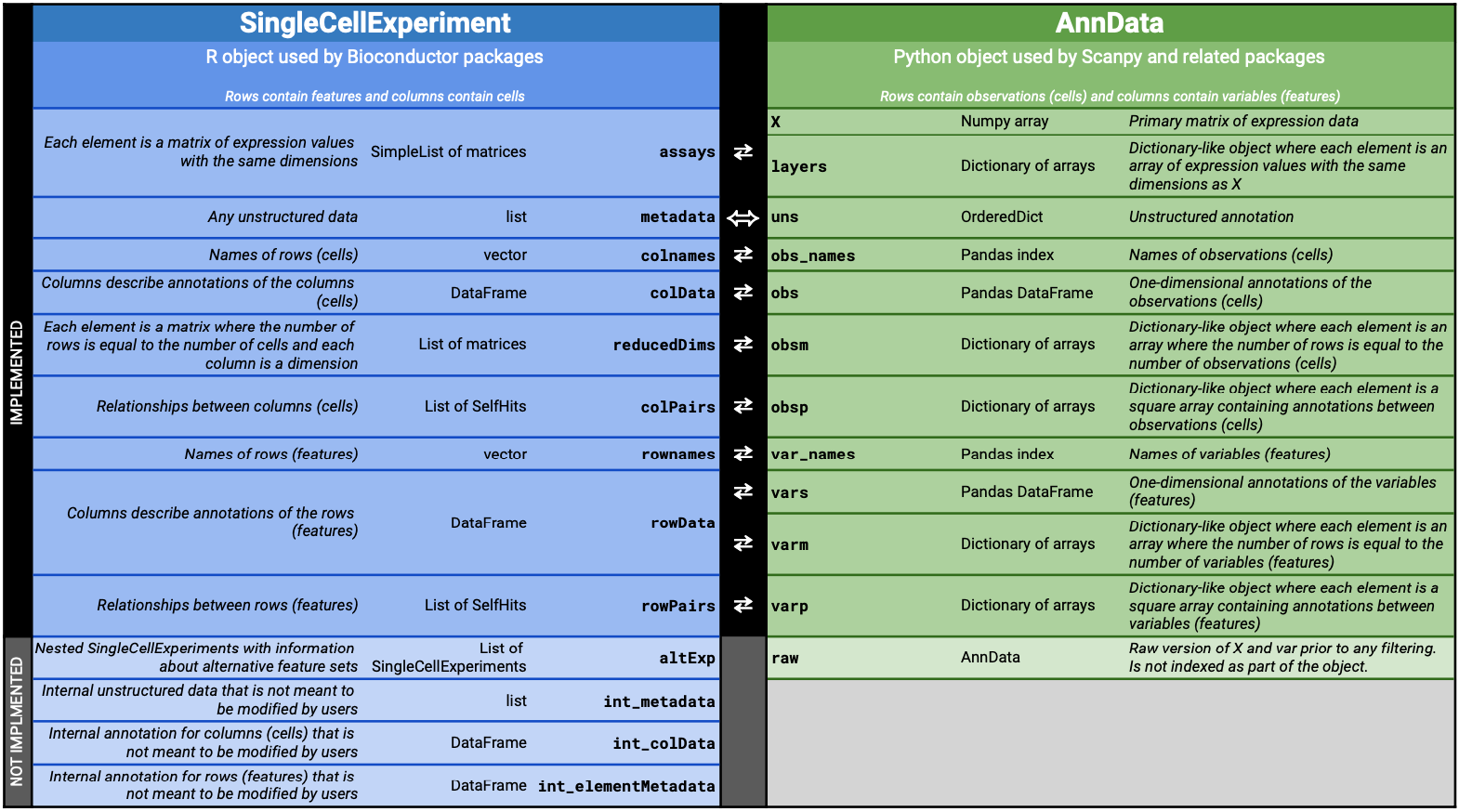
Matrix conversion
In SCE2AnnData(), matrices are converted to a numpy-friendly format.
Sparse matrices are converted to dgCMatrix
objects while all other matrices are converted into ordinary matrices. If
skip_assays = TRUE, empty sparse matrices are created instead and the user
is expected to fill in the assays on the Python side.
For AnnData2SCE(), a warning is raised if there is no corresponding R
format for a matrix in the AnnData object, and an empty sparse matrix is
created instead as a placeholder. If skip_assays = NA, no warning is
emitted but variables are created in the
int_metadata of the
output to specify which assays were skipped.
If skip_assays = TRUE, empty sparse matrices are created for all assays,
regardless of whether they might be convertible to an R format or not.
In both cases, the user is expected to fill in the assays on the R side.
metadata/uns conversion
We attempt to convert between items in the
SingleCellExperiment::SingleCellExperiment
metadata() slot and the AnnData uns slot. If
an item cannot be converted a warning will be raised.
varm conversion
Values stored in the varm slot of an AnnData object are stored in a
column of rowData() in a
SingleCellExperiment::SingleCellExperiment as a
DataFrame of matrices. If this column is
present an attempt is made to transfer this information when converting
from SingleCellExperiment::SingleCellExperiment to AnnData.
SpatialExperiment conversion
In SCE2AnnData(), if sce is a SpatialExperiment::SpatialExperiment
object, the spatial coordinates are added to the reducedDims slot before
conversion to an AnnData object.
See also
writeH5AD() and readH5AD() for dealing directly with H5AD files.
Examples
if (requireNamespace("scRNAseq", quietly = TRUE)) {
library(basilisk)
library(scRNAseq)
seger <- SegerstolpePancreasData()
# These functions are designed to be run inside
# a specified Python environment
roundtrip <- basiliskRun(fun = function(sce) {
# Convert SCE to AnnData:
adata <- zellkonverter::SCE2AnnData(sce)
# Maybe do some work in Python on 'adata':
# BLAH BLAH BLAH
# Convert back to an SCE:
zellkonverter::AnnData2SCE(adata)
}, env = zellkonverterAnnDataEnv(), sce = seger)
}
#> Loading required package: reticulate
#> Loading required package: SingleCellExperiment
#> Loading required package: SummarizedExperiment
#> Loading required package: MatrixGenerics
#> Loading required package: matrixStats
#>
#> Attaching package: 'MatrixGenerics'
#> The following objects are masked from 'package:matrixStats':
#>
#> colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
#> colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
#> colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
#> colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
#> colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
#> colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
#> colWeightedMeans, colWeightedMedians, colWeightedSds,
#> colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
#> rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
#> rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
#> rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
#> rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
#> rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
#> rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
#> rowWeightedSds, rowWeightedVars
#> Loading required package: GenomicRanges
#> Loading required package: stats4
#> Loading required package: BiocGenerics
#> Loading required package: generics
#>
#> Attaching package: 'generics'
#> The following objects are masked from 'package:base':
#>
#> as.difftime, as.factor, as.ordered, intersect, is.element, setdiff,
#> setequal, union
#>
#> Attaching package: 'BiocGenerics'
#> The following objects are masked from 'package:stats':
#>
#> IQR, mad, sd, var, xtabs
#> The following objects are masked from 'package:base':
#>
#> Filter, Find, Map, Position, Reduce, anyDuplicated, aperm, append,
#> as.data.frame, basename, cbind, colnames, dirname, do.call,
#> duplicated, eval, evalq, get, grep, grepl, is.unsorted, lapply,
#> mapply, match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
#> rank, rbind, rownames, sapply, saveRDS, table, tapply, unique,
#> unsplit, which.max, which.min
#> Loading required package: S4Vectors
#>
#> Attaching package: 'S4Vectors'
#> The following object is masked from 'package:utils':
#>
#> findMatches
#> The following objects are masked from 'package:base':
#>
#> I, expand.grid, unname
#> Loading required package: IRanges
#> Loading required package: Seqinfo
#> Loading required package: Biobase
#> Welcome to Bioconductor
#>
#> Vignettes contain introductory material; view with
#> 'browseVignettes()'. To cite Bioconductor, see
#> 'citation("Biobase")', and for packages 'citation("pkgname")'.
#>
#> Attaching package: 'Biobase'
#> The following object is masked from 'package:MatrixGenerics':
#>
#> rowMedians
#> The following objects are masked from 'package:matrixStats':
#>
#> anyMissing, rowMedians
#> Installing pyenv ...
#> Done! pyenv has been installed to '/github/home/.local/share/r-reticulate/pyenv/bin/pyenv'.
#> Using Python: /github/home/.pyenv/versions/3.14.0/bin/python3.14
#> Creating virtual environment '/github/home/.cache/R/basilisk/1.25.0/zellkonverter/1.23.0/zellkonverterAnnDataEnv-0.12.3' ...
#> + /github/home/.pyenv/versions/3.14.0/bin/python3.14 -m venv /github/home/.cache/R/basilisk/1.25.0/zellkonverter/1.23.0/zellkonverterAnnDataEnv-0.12.3
#> Done!
#> Installing packages: pip, wheel, setuptools
#> + /github/home/.cache/R/basilisk/1.25.0/zellkonverter/1.23.0/zellkonverterAnnDataEnv-0.12.3/bin/python -m pip install --upgrade pip wheel setuptools
#> Installing packages: 'anndata==0.12.3', 'h5py==3.15.1', 'natsort==8.4.0', 'numpy==2.3.4', 'pandas==2.3.3', 'scipy==1.16.2'
#> + /github/home/.cache/R/basilisk/1.25.0/zellkonverter/1.23.0/zellkonverterAnnDataEnv-0.12.3/bin/python -m pip install --upgrade --no-user 'anndata==0.12.3' 'h5py==3.15.1' 'natsort==8.4.0' 'numpy==2.3.4' 'pandas==2.3.3' 'scipy==1.16.2'
#> Virtual environment '/github/home/.cache/R/basilisk/1.25.0/zellkonverter/1.23.0/zellkonverterAnnDataEnv-0.12.3' successfully created.
#> For native R and reading and writing of H5AD files, an R <AnnData> object, and
#> conversion to <SingleCellExperiment> or <Seurat> objects, check out the
#> anndataR package:
#> ℹ Install it from Bioconductor with `BiocManager::install("anndataR")`
#> ℹ See more at <https://bioconductor.org/packages/anndataR/>
#> This message is displayed once per session.
#> ℹ Using the 'counts' assay as the X matrix
#> ℹ Using stored X_name value 'counts'
#> Warning: index contains duplicated values: row names not set
#> Warning: The names of these selected obs columns have been modified to match R
#> conventions: 'body mass index' -> 'body.mass.index', 'clinical information' ->
#> 'clinical.information', 'single cell well quality' ->
#> 'single.cell.well.quality', 'submitted single cell quality' ->
#> 'submitted.single.cell.quality', and 'cell type' -> 'cell.type'