Intestinal organoid differentiation - Data preprocessing#
Preprocessing of scEU-seq organoid data by
Excluding control cells identified via labeling time
"dmso"Excluding tuft cells
Removing genes with less than 50 counts
Normalizing counts
Extracting 2000 highly variable features
Log1p transforming total counts stored in
adata.XComputing PCA
Constructing a nearest neighbor graph with 30 neighbors and 30 principal components
Computing UMAP embedding
Library imports#
import os
import sys
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
import scvelo as scv
from cr2 import running_in_notebook
sys.path.extend(["../../", "."])
from paths import DATA_DIR, FIG_DIR # isort: skip # noqa: E402
Global seed set to 0
General settings#
sc.settings.verbosity = 3
scv.settings.verbosity = 3
scv.settings.set_figure_params("scvelo")
SAVE_FIGURES = True
if SAVE_FIGURES:
os.makedirs(FIG_DIR / "labeling_kernel", exist_ok=True)
Data loading#
adata = sc.read(DATA_DIR / "sceu_organoid" / "processed" / "raw.h5ad")
adata = adata[adata.obs["labeling_time"] != "dmso", :].copy()
adata = adata[~adata.obs["cell_type"].isin(["Tuft cells"]), :]
adata.obs["labeling_time"] = adata.obs["labeling_time"].astype(float) / 60
adata.obs.drop(["well_id", "batch_id", "log10_gfp", "monocle_branch_id", "monocle_pseudotime"], axis=1, inplace=True)
del (
adata.layers["labeled_unspliced"],
adata.layers["labeled_spliced"],
adata.layers["unlabeled_unspliced"],
adata.layers["unlabeled_spliced"],
)
adata
AnnData object with n_obs × n_vars = 3452 × 9157
obs: 'experiment', 'labeling_time', 'cell_type', 'som_cluster_id'
var: 'ensum_id'
uns: 'cell_type_colors'
obsm: 'X_umap_paper'
layers: 'labeled', 'total', 'unlabeled'
pd.DataFrame(adata.obs[["labeling_time", "experiment"]].groupby("experiment").apply(lambda x: x.value_counts())).rename(
{0: "value_counts"}, axis=1
).droplevel(level=2).sort_index()
| value_counts | ||
|---|---|---|
| experiment | labeling_time | |
| Chase | 0.00 | 647 |
| 0.75 | 805 | |
| 6.00 | 632 | |
| Pulse | 2.00 | 1368 |
adata.obs["cell_type_merged"] = adata.obs["cell_type"].copy()
adata.obs["cell_type_merged"].replace({"Enteroendocrine cells": "Enteroendocrine progenitors"}, inplace=True)
Data preprocessing#
scv.pp.filter_and_normalize(
adata, min_counts=50, layers_normalize=["X", "labeled", "unlabeled", "total"], n_top_genes=2000
)
adata
WARNING: Could not find spliced / unspliced counts.
Normalized count data: X, labeled, unlabeled, total.
Extracted 2000 highly variable genes.
Logarithmized X.
AnnData object with n_obs × n_vars = 3452 × 2000
obs: 'experiment', 'labeling_time', 'cell_type', 'som_cluster_id', 'cell_type_merged', 'initial_size', 'n_counts'
var: 'ensum_id', 'gene_count_corr', 'means', 'dispersions', 'dispersions_norm', 'highly_variable'
uns: 'cell_type_colors'
obsm: 'X_umap_paper'
layers: 'labeled', 'total', 'unlabeled'
sc.tl.pca(adata, svd_solver="arpack")
sc.pp.neighbors(adata, n_neighbors=30, n_pcs=30)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:01)
computing neighbors
using 'X_pca' with n_pcs = 30
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:05)
sc.tl.umap(adata)
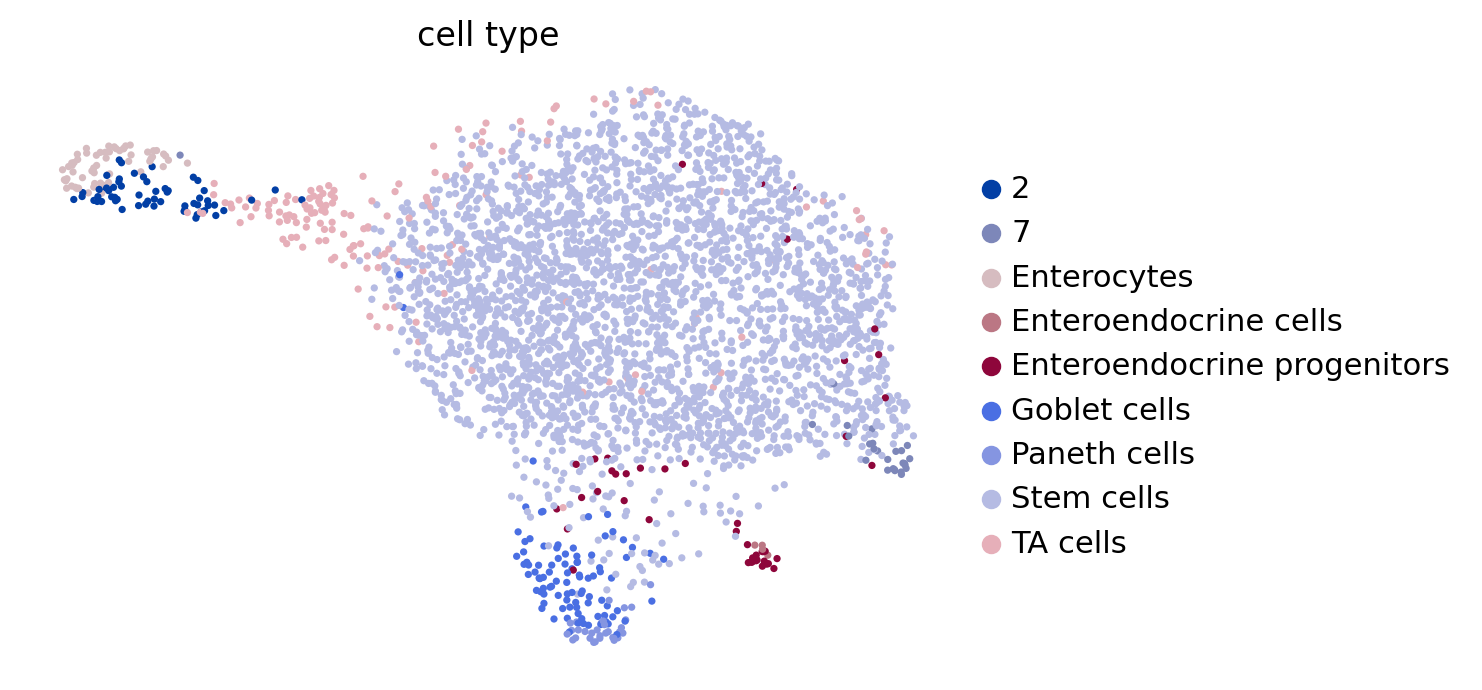
if running_in_notebook():
scv.pl.scatter(adata, basis="umap", color="cell_type", legend_loc="right")
if SAVE_FIGURES:
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(adata, basis="umap", color="cell_type", title="", legend_loc=False, show=False, ax=ax)
fig.savefig(
FIG_DIR / "labeling_kernel" / "umap_colored_by_cell_type.eps",
format="eps",
transparent=True,
bbox_inches="tight",
)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:05)

filepath = DATA_DIR / "sceu_organoid" / "processed" / "umap_coords.csv"
pd.DataFrame(adata.obsm["X_umap"], index=adata.obs_names, columns=["umap_1", "umap_2"]).to_csv(filepath)
Data saving#
adata.write(DATA_DIR / "sceu_organoid" / "processed" / "preprocessed.h5ad")