Pharyngeal endoderm development analysis with WOT#
Import packages#
import os
import sys
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as sc
import scvelo as scv
import wot
from cr2 import running_in_notebook
sys.path.extend(["../../../", "."])
from paths import DATA_DIR, FIG_DIR # isort: skip # noqa: E402
Global seed set to 0
General settings#
sc.settings.verbosity = 2
scv.settings.verbosity = 3
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=20, color_map="viridis")
SAVE_FIGURES = False
if SAVE_FIGURES:
os.makedirs(FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm", exist_ok=True)
SHOW_COLORBAR = not SAVE_FIGURES
(DATA_DIR / "pharyngeal_endoderm" / "results").mkdir(parents=True, exist_ok=True)
Constants#
# fmt: off
S_GENES = [
"Mcm5", "Pcna", "Tyms", "Fen1", "Mcm2", "Mcm4", "Rrm1", "Ung", "Gins2",
"Mcm6", "Cdca7", "Dtl", "Prim1", "Uhrf1", "Mlf1ip", "Hells", "Rfc2",
"Rpa2", "Nasp", "Rad51ap1", "Gmnn", "Wdr76", "Slbp", "Ccne2", "Ubr7",
"Pold3", "Msh2", "Atad2", "Rad51", "Rrm2", "Cdc45", "Cdc6", "Exo1",
"Tipin", "Dscc1", "Blm", "Casp8ap2", "Usp1", "Clspn", "Pola1", "Chaf1b",
"Brip1", "E2f8",
]
G2M_GENES = [
"Hmgb2", "Cdk1", "Nusap1", "Ube2c", "Birc5", "Tpx2", "Top2a", "Ndc80",
"Cks2", "Nuf2", "Cks1b", "Mki67", "Tmpo", "Cenpf", "Tacc3", "Fam64a",
"Smc4", "Ccnb2", "Ckap2l", "Ckap2", "Aurkb", "Bub1", "Kif11", "Anp32e",
"Tubb4b", "Gtse1", "Kif20b", "Hjurp", "Cdca3", "Hn1", "Cdc20", "Ttk",
"Cdc25c", "Kif2c", "Rangap1", "Ncapd2", "Dlgap5", "Cdca2", "Cdca8",
"Ect2", "Kif23", "Hmmr", "Aurka", "Psrc1", "Anln", "Lbr", "Ckap5",
"Cenpe", "Ctcf", "Nek2", "G2e3", "Gas2l3", "Cbx5", "Cenpa",
]
# fmt: on
TERMINAL_STATES = ["mTEC", "cTEC", "parathyroid", "ubb"]
Data loading#
adata = sc.read(DATA_DIR / "pharyngeal_endoderm" / "raw" / "adata_pharynx.h5ad")
adata.obsm["X_umap"] = adata.obs[["UMAP1", "UMAP2"]].values
adata.obs["day"] = adata.obs["day_str"].astype(float)
adata.obs = adata.obs[["cluster_name", "day", "is_doublet"]]
adata.obs["cluster_fine"] = (
pd.read_csv(DATA_DIR / "pharyngeal_endoderm" / "raw" / "cluster_data.csv", index_col=0)
.loc[adata.obs_names, "res.1"]
.values
)
adata.obs["cluster_fine"] = adata.obs["cluster_fine"].astype(str).astype("category")
adata = adata[adata.obs["cluster_fine"].isin(["2", "4", "9", "12", "25", "26"]), :].copy()
adata.uns["cluster_name_colors"] = ["#023fa5", "#bec1d4", "#b5bbe3", "#e07b91", "#11c638"]
adata
AnnData object with n_obs × n_vars = 11073 × 27998
obs: 'cluster_name', 'day', 'is_doublet', 'cluster_fine'
uns: 'cluster_name_colors'
obsm: 'X_umap'
mouse_tfs = (
pd.read_csv(DATA_DIR / "generic" / "mouse_tfs.tsv", sep="\t", header=None)
.rename(columns={0: "Ensemble ID", 1: "Gene ID"})["Gene ID"]
.tolist()
)
Data preprocessing#
sc.pp.highly_variable_genes(adata)
extracting highly variable genes
finished (0:00:03)
sc.tl.pca(adata)
sc.pp.neighbors(adata, n_pcs=30, n_neighbors=30)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:00)
computing neighbors
using 'X_pca' with n_pcs = 30
finished (0:02:28)
sc.tl.umap(adata)
computing UMAP
finished (0:00:06)
if running_in_notebook():
scv.pl.scatter(
adata, basis="umap", c="cluster_name", title="", dpi=250, legend_fontsize=12, legend_fontweight="normal"
)

WOT#
if not (DATA_DIR / "pharyngeal_endoderm" / "tmaps_subsetted_data").exists():
ot_model = wot.ot.OTModel(adata)
ot_model.compute_all_transport_maps(tmap_out=DATA_DIR / "pharyngeal_endoderm" / "tmaps_subsetted_data" / "tmaps")
cell_sets = {
terminal_state: adata.obs_names[
adata.obs["cluster_name"].isin([terminal_state]) & adata.obs["day"].isin([12.5])
].tolist()
for terminal_state in TERMINAL_STATES
}
tmap_model = wot.tmap.TransportMapModel.from_directory(
DATA_DIR / "pharyngeal_endoderm" / "tmaps_subsetted_data" / "tmaps"
)
populations = tmap_model.population_from_cell_sets(cell_sets, at_time=12.5)
res = tmap_model.pull_back(populations[0], to_time=10.5)
trajectory_ds = tmap_model.trajectories(populations)
for cell_type in TERMINAL_STATES:
adata.obs[f"traj_{cell_type}"] = trajectory_ds[:, cell_type].X.squeeze().copy()
if running_in_notebook():
scv.pl.scatter(
adata,
basis="umap",
c=[f"traj_{terminal_state}" for terminal_state in TERMINAL_STATES],
cmap="viridis",
title="",
legend_loc=None,
)
if SAVE_FIGURES:
for terminal_state in TERMINAL_STATES:
fig, ax = plt.subplots(figsize=(6, 4))
if running_in_notebook():
scv.pl.scatter(
adata,
basis="umap",
color=f"traj_{terminal_state}",
s=50,
cmap="viridis",
title="",
colorbar=SHOW_COLORBAR,
ax=ax,
)
fig.savefig(
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"wot_pullback_{terminal_state}_subsetted_data.eps",
format="eps",
transparent=True,
bbox_inches="tight",
)
if running_in_notebook():
scv.pl.scatter(
adata,
basis="umap",
c=[f"traj_{terminal_state}" for terminal_state in TERMINAL_STATES],
cmap="viridis",
# title="",
legend_loc=None,
perc=[0, 99],
)
if SAVE_FIGURES:
for terminal_state in TERMINAL_STATES:
fig, ax = plt.subplots(figsize=(6, 4))
if running_in_notebook():
scv.pl.scatter(
adata,
basis="umap",
color=f"traj_{terminal_state}",
s=50,
cmap="viridis",
title="",
colorbar=SHOW_COLORBAR,
perc=[0, 99],
ax=ax,
)
fig.savefig(
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"wot_pullback_{terminal_state}_clipped_subsetted_data.eps",
format="eps",
transparent=True,
bbox_inches="tight",
)
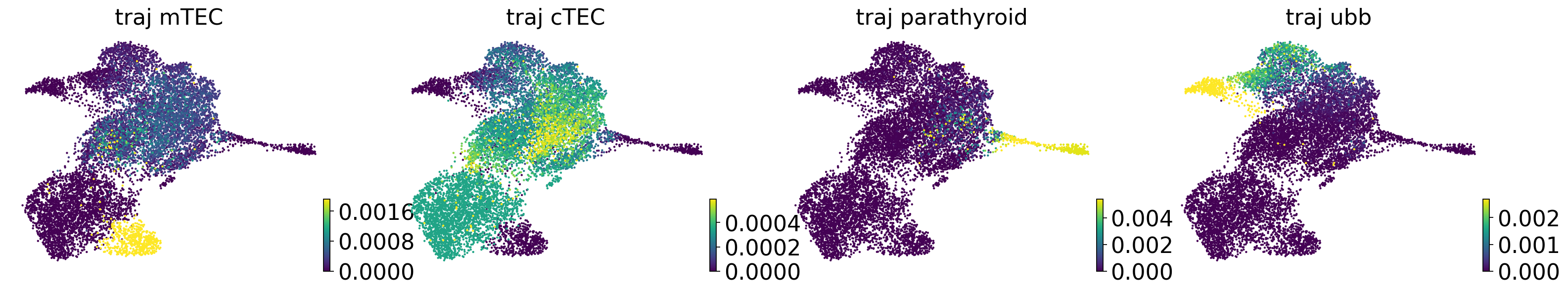
Driver analysis#
# fmt: off
mtec_genes = [
"Cldn3", "Cldn4", "Notch1", "Krt5", "H2-Aa", "H2-Ab1", "H2-Eb1",
"Grhl3", "Grhl1", "Elf5", "Irf6", "Sox9", "Upk2", "Ovol1", "Hes1",
"Rhov", "Pvrl4", "Klf5", "Egr1", "Sfn", "Perp", "Fxyd3", "Hspb1",
"Krt5", "S100a11",
]
# fmt: on
target_cell_set = tmap_model.population_from_cell_sets({"mTEC": cell_sets["mTEC"]}, at_time=12.5)
fate_ds = tmap_model.fates(target_cell_set)
results = {}
gene_names = {}
genes_to_exclude = adata.var_names[
adata.var_names.str.startswith(("mt.", "Rpl", "Rps", "^Hb[^(p)]")) | adata.var_names.isin(S_GENES + G2M_GENES)
]
for day in [9.5, 10.5, 11.5]:
results[day] = wot.tmap.diff_exp(adata[adata.obs["day"].isin([day])], fate_ds, compare="all")
gene_names[day] = (
results[day][(results[day]["t_fdr"] < 0.01) & (results[day]["name1"] == "mTEC")]
.sort_values("t_score", ascending=False)
.index
)
gene_names[day] = gene_names[day][~gene_names[day].isin(genes_to_exclude)]
if len(gene_names[day]) > 0:
df = pd.DataFrame(
results[day].loc[gene_names[day][:20], "t_score"],
)
df["ranking"] = np.arange(20)
fig, ax = plt.subplots(figsize=(6, 4))
y_min = np.min(df["t_score"])
y_max = np.max(df["t_score"])
y_min -= 0.1 * (y_max - y_min)
y_max += 0.4 * (y_max - y_min)
# ax = fig.add_subplot(gs[count])
ax.set_ylim(y_min, y_max)
ax.set_xlim(-0.75, 19.5)
for gene in df.index:
if gene in mouse_tfs:
color = "#FEAE00"
elif gene in mtec_genes:
color = "#00AB8E"
else:
color = "#000000"
ax.text(
df.loc[gene, "ranking"],
df.loc[gene, "t_score"],
gene,
rotation="vertical",
verticalalignment="bottom",
horizontalalignment="center",
fontsize=20,
color=color,
)
if SAVE_FIGURES:
fig.savefig(
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"genes_ranked_by_t_score_original_wot_day_{day}.eps",
format="eps",
transparent=True,
bbox_inches="tight",
)
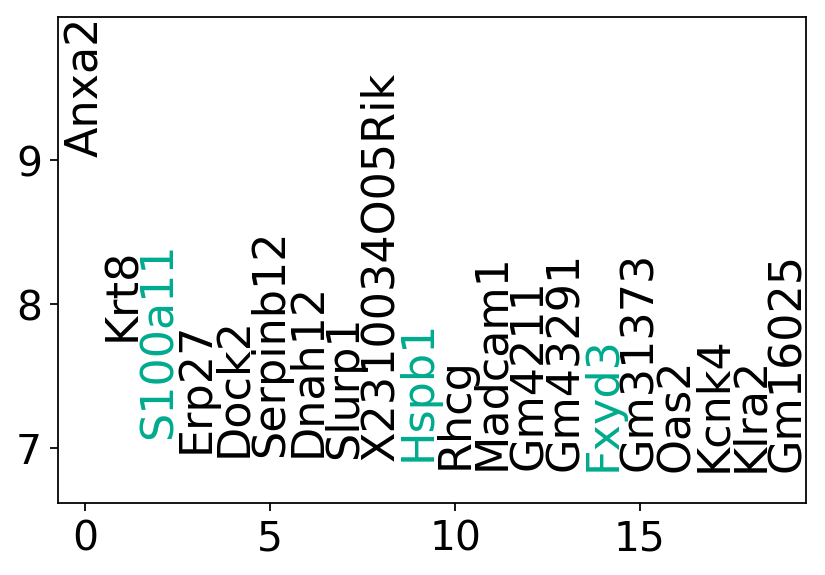
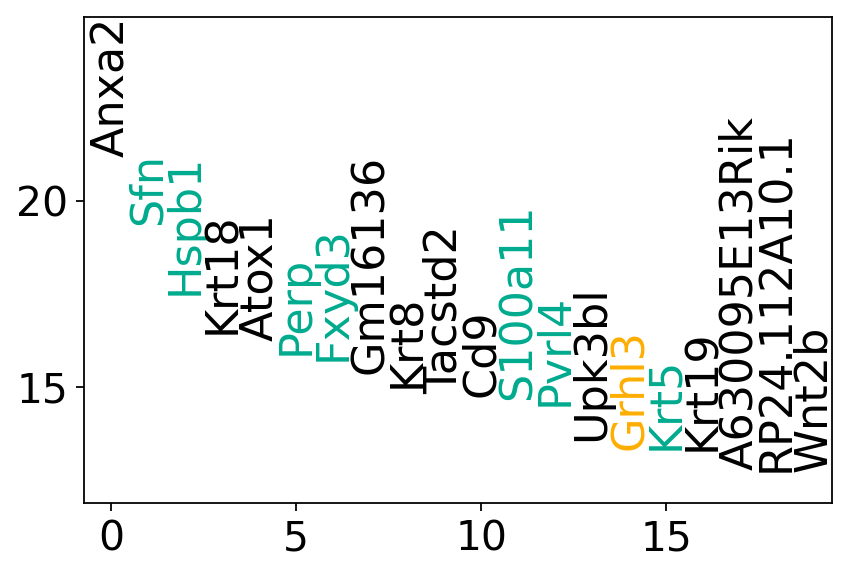
for day, genes in gene_names.items():
gene_is_tf = genes[:100].isin(mouse_tfs)
n_tfs = gene_is_tf.sum()
print(f"Number of TFs in first 100 genes at day {day}: {n_tfs}")
gene_is_mtec_marker = genes[:100].isin(mtec_genes)
n_mtec_genes = gene_is_mtec_marker.sum()
print(f"Number of mTEC genes in first 100 genes at day {day}: {n_mtec_genes}\n")
Number of TFs in first 100 genes at day 9.5: 0
Number of mTEC genes in first 100 genes at day 9.5: 0
Number of TFs in first 100 genes at day 10.5: 2
Number of mTEC genes in first 100 genes at day 10.5: 3
Number of TFs in first 100 genes at day 11.5: 4
Number of mTEC genes in first 100 genes at day 11.5: 8
cr_ranking = pd.read_csv(DATA_DIR / "pharyngeal_endoderm" / "results" / "driver_genes_ranking_cr.csv", index_col=0)
_mtec_genes = list(set(mtec_genes).intersection(cr_ranking.index))
for day in [9.5, 10.5, 11.5]:
wot_ranking = (
results[day][results[day]["name1"] == "mTEC"].sort_values("t_score", ascending=False)["t_score"].to_frame()
)
wot_ranking["ranking"] = np.arange(wot_ranking.shape[0])
_perc = (cr_ranking.loc[_mtec_genes, "ranking"] <= wot_ranking.loc[_mtec_genes, "ranking"]).mean() * 100
print(f"Percentage of genes that CR ranks higher or equal than classical DE (day {day}): {_perc:.2f}%")
_perc = (cr_ranking.loc[_mtec_genes, "ranking"] < wot_ranking.loc[_mtec_genes, "ranking"]).mean() * 100
print(f"Percentage of genes that CR ranks strictly higher than classical DE (day {day}): {_perc:.2f}%\n")
Percentage of genes that CR ranks higher or equal than classical DE (day 9.5): 95.24%
Percentage of genes that CR ranks strictly higher than classical DE (day 9.5): 95.24%
Percentage of genes that CR ranks higher or equal than classical DE (day 10.5): 85.71%
Percentage of genes that CR ranks strictly higher than classical DE (day 10.5): 85.71%
Percentage of genes that CR ranks higher or equal than classical DE (day 11.5): 57.14%
Percentage of genes that CR ranks strictly higher than classical DE (day 11.5): 52.38%