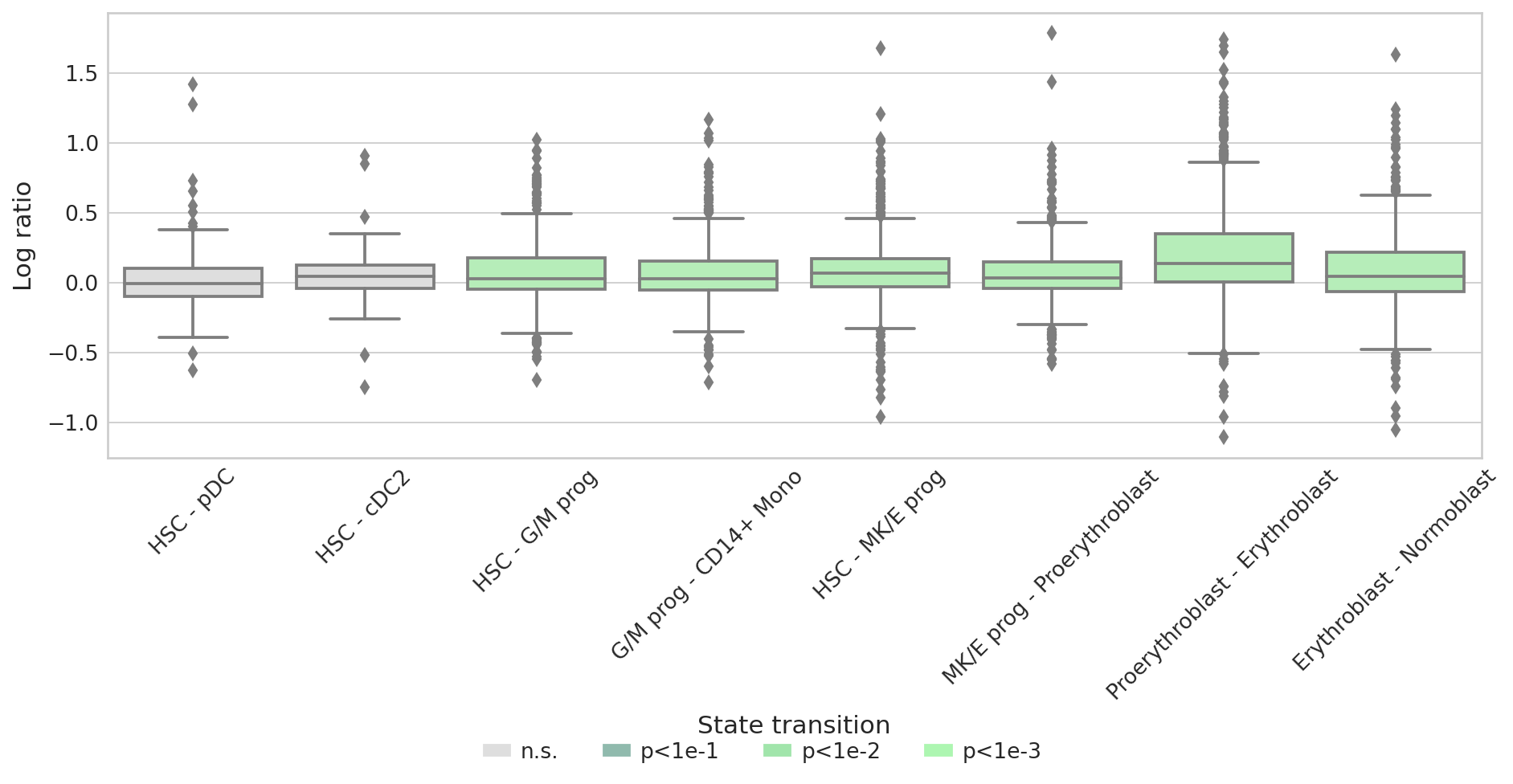
Cross boundary correctness score#
Analysis of cross boundary correctness (CBC) score of pseudotime-based analysis with the PseudotimeKernel and an RNA velocity-based analysis with VelocityKernel on the NeurIPS 2021 hematopoiesis data.
Library imports#
import os
import sys
from tqdm import tqdm
import numpy as np
import pandas as pd
from scipy.stats import ttest_ind
import matplotlib.pyplot as plt
import mplscience
import seaborn as sns
from matplotlib.patches import Patch
import cellrank as cr
import scanpy as sc
import scvelo as scv
from anndata import AnnData
from cr2 import running_in_notebook
sys.path.extend(["../../../", "."])
from paths import DATA_DIR, FIG_DIR # isort: skip # noqa: E402
Global seed set to 0
General settings#
sc.settings.verbosity = 2
cr.settings.verbosity = 4
scv.settings.verbosity = 3
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=20, color_map="viridis")
SAVE_FIGURES = False
if SAVE_FIGURES:
os.makedirs(FIG_DIR / "pseudotime_kernel" / "hematopoiesis", exist_ok=True)
FIGURE_FORMAT = "pdf"
os.makedirs(DATA_DIR / "hematopoiesis" / "results", exist_ok=True)
Constants#
N_JOBS = 8
CELLTYPES_TO_KEEP = [
"HSC",
"MK/E prog",
"Proerythroblast",
"Erythroblast",
"Normoblast",
"cDC2",
"pDC",
"G/M prog",
"CD14+ Mono",
]
STATE_TRANSITIONS = [
("HSC", "pDC"),
("HSC", "cDC2"),
("HSC", "G/M prog"),
("G/M prog", "CD14+ Mono"),
("HSC", "MK/E prog"),
("MK/E prog", "Proerythroblast"),
("Proerythroblast", "Erythroblast"),
("Erythroblast", "Normoblast"),
]
Function definitions#
def get_significance(pvalue) -> str:
"""Assign significance symbol based on p-value."""
if pvalue < 0.001:
return "***"
elif pvalue < 0.01:
return "**"
elif pvalue < 0.1:
return "*"
else:
return "n.s."
def get_dpt_adata() -> AnnData:
"""Load and preprocess data for pseudotime-based analysis."""
adata = sc.read(DATA_DIR / "hematopoiesis" / "processed" / "gex_preprocessed.h5ad")
adata = adata[adata.obs["l2_cell_type"].isin(CELLTYPES_TO_KEEP), :].copy()
sc.pp.neighbors(adata, use_rep="MultiVI_latent")
sc.tl.umap(adata)
sc.tl.diffmap(adata, n_comps=15)
df = (
pd.DataFrame(
{
"diff_comp": adata.obsm["X_diffmap"][:, 5],
"cell_type": adata.obs["l2_cell_type"].values,
}
)
.reset_index()
.rename({"index": "obs_id"}, axis=1)
)
df = df.loc[df["cell_type"] == "HSC", "diff_comp"]
root_idx = df.index[df.argmax()]
adata.uns["iroot"] = root_idx
sc.tl.dpt(adata, n_dcs=6)
return adata
def get_velo_adata() -> AnnData:
"""Load and preprocess data for RNA velocity-based analysis."""
adata = sc.read(DATA_DIR / "hematopoiesis" / "processed" / "gex_velocity.h5ad")
adata = adata[adata.obs["l2_cell_type"].isin(CELLTYPES_TO_KEEP), :].copy()
scv.pp.filter_genes(adata, min_shared_counts=20)
scv.pp.normalize_per_cell(adata)
sc.pp.neighbors(adata, use_rep="MultiVI_latent")
sc.tl.umap(adata)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
scv.tl.recover_dynamics(adata, n_jobs=N_JOBS)
scv.tl.velocity(adata, mode="dynamical")
return adata
Data loading#
adatas = {}
adatas["dpt"] = get_dpt_adata()
adatas["dpt"].obs["obs_id"] = np.arange(0, adatas["dpt"].n_obs)
adatas["dpt"]
computing neighbors
finished (0:02:26)
computing UMAP
finished (0:00:10)
computing Diffusion Maps using n_comps=15(=n_dcs)
computing transitions
finished (0:00:00)
eigenvalues of transition matrix
[1. 0.99922997 0.9977195 0.9968419 0.9955766 0.9942717
0.9900949 0.9884704 0.9867782 0.9852537 0.9849594 0.9830871
0.98229724 0.9809607 0.97756666]
finished (0:00:00)
computing Diffusion Pseudotime using n_dcs=6
finished (0:00:00)
AnnData object with n_obs × n_vars = 24440 × 25629
obs: 'site', 'donor', 'batch', 'l1_cell_type', 'l2_cell_type', 'dpt_pseudotime', 'obs_id'
var: 'hvg_multiVI'
uns: 'neighbors', 'umap', 'diffmap_evals', 'iroot'
obsm: 'MultiVI_latent', 'X_umap', 'X_diffmap'
layers: 'counts'
obsp: 'connectivities', 'distances'
adatas["rna_velocity"] = get_velo_adata()
adatas["rna_velocity"]
Filtered out 16169 genes that are detected 20 counts (shared).
WARNING: Did not normalize X as it looks processed already. To enforce normalization, set `enforce=True`.
Normalized count data: spliced, unspliced.
computing neighbors
finished (0:00:02)
computing UMAP
finished (0:00:10)
computing moments based on connectivities
finished (0:00:09) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
recovering dynamics (using 8/14 cores)
Global seed set to 0
Global seed set to 0
Global seed set to 0
Global seed set to 0
Global seed set to 0
Global seed set to 0
Global seed set to 0
Global seed set to 0
finished (0:03:29) --> added
'fit_pars', fitted parameters for splicing dynamics (adata.var)
computing velocities
finished (0:00:19) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
AnnData object with n_obs × n_vars = 24440 × 9460
obs: 'batch', 'site', 'donor', 'l1_cell_type', 'l2_cell_type', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts'
var: 'hvg_multiVI', 'fit_r2', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_variance', 'fit_alignment_scaling', 'velocity_genes'
uns: 'neighbors', 'umap', 'recover_dynamics', 'velocity_params'
obsm: 'MultiVI_latent', 'X_umap'
varm: 'loss'
layers: 'counts', 'spliced', 'unspliced', 'Ms', 'Mu', 'fit_t', 'fit_tau', 'fit_tau_', 'velocity', 'velocity_u'
obsp: 'connectivities', 'distances'
CellRank analysis#
Kernel#
ptk = cr.kernels.PseudotimeKernel(adatas["dpt"], time_key="dpt_pseudotime").compute_transition_matrix(
threshold_scheme="soft"
)
vk = cr.kernels.VelocityKernel(adatas["rna_velocity"]).compute_transition_matrix()
ck = cr.kernels.ConnectivityKernel(adatas["rna_velocity"]).compute_transition_matrix()
vk_ck = 0.2 * ck + 0.8 * vk
kernels = {"PseudotimeKernel": ptk, "VelocityKernel": vk_ck}
Computing transition matrix based on pseudotime
Finish (0:00:10)
Computing transition matrix using `'deterministic'` model
Using `softmax_scale=10.3350`
Finish (0:00:19)
Computing transition matrix based on `adata.obsp['connectivities']`
DEBUG: Density normalizing the transition matrix
Finish (0:00:00)
Cross-boundary correctness score#
cluster_key = "l2_cell_type"
rep = "MultiVI_latent"
score_df = []
for source, target in tqdm(STATE_TRANSITIONS):
cbc_ptk = ptk.cbc(source=source, target=target, cluster_key=cluster_key, rep=rep)
cbc_velo = vk_ck.cbc(source=source, target=target, cluster_key=cluster_key, rep=rep)
score_df.append(
pd.DataFrame(
{
"State transition": [f"{source} - {target}"] * len(cbc_ptk),
"Log ratio": np.log((cbc_ptk + 1) / (cbc_velo + 1)),
}
)
)
score_df = pd.concat(score_df)
100%|██████████| 8/8 [00:07<00:00, 1.02it/s]
dfs = []
ttest_res = {}
significances = {}
for source, target in STATE_TRANSITIONS:
obs_mask = score_df["State transition"].isin([f"{source} - {target}"])
a = score_df.loc[obs_mask, "Log ratio"].values
b = np.zeros(len(a))
ttest_res[f"{source} - {target}"] = ttest_ind(a, b, equal_var=False, alternative="greater")
significances[f"{source} - {target}"] = get_significance(ttest_res[f"{source} - {target}"].pvalue)
significance_palette = {"n.s.": "#dedede", "*": "#90BAAD", "**": "#A1E5AB", "***": "#ADF6B1"}
palette = {
state_transition: significance_palette[significance] for state_transition, significance in significances.items()
}
if running_in_notebook():
with mplscience.style_context():
sns.set_style(style="whitegrid")
fig, ax = plt.subplots(figsize=(12, 6))
sns.boxplot(data=score_df, x="State transition", y="Log ratio", palette=palette, ax=ax)
ax.tick_params(axis="x", rotation=45)
handles = [Patch(label=label, facecolor=color) for label, color in significance_palette.items()]
fig.legend(
handles=handles,
labels=["n.s.", "p<1e-1", "p<1e-2", "p<1e-3"],
loc="lower center",
ncol=4,
bbox_to_anchor=(0.5, -0.025),
)
fig.tight_layout()
plt.show()
if SAVE_FIGURES:
ax.set(xlabel="", xticklabels="", ylabel="", yticklabels="")
fig.legends = []
fig.savefig(
FIG_DIR / "pseudotime_kernel" / "hematopoiesis" / f"log_ratio_cross_boundary.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)