Pharyngeal endoderm development analysis with the RealTimeKernel#
Import packages#
import sys
import pandas as pd
import matplotlib.pyplot as plt
import mplscience
import seaborn as sns
import cellrank as cr
import scanpy as sc
import scvelo as scv
from anndata import AnnData
from cr2 import running_in_notebook
sys.path.extend(["../../../", "."])
from paths import DATA_DIR, FIG_DIR # isort: skip # noqa: E402
Global seed set to 0
General settings#
sc.settings.verbosity = 2
cr.settings.verbosity = 4
scv.settings.verbosity = 3
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=20, color_map="viridis")
SAVE_FIGURES = False
if SAVE_FIGURES:
(FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm").mkdir(parents=True, exist_ok=True)
FIGURE_FORMAT = "pdf"
(DATA_DIR / "pharyngeal_endoderm" / "processed").mkdir(parents=True, exist_ok=True)
N_JOBS = 8
Constants#
Data loading#
adata = sc.read(DATA_DIR / "pharyngeal_endoderm" / "processed" / "adata_velo.h5ad")
adata.obs["cluster_name"] = (
adata.obs["cluster_name"]
.astype(str)
.astype("category")
.cat.rename_categories({"nan": "progenitors"})
.cat.reorder_categories(["progenitors"] + adata.obs["cluster_name"].cat.categories.tolist())
)
adata.uns["cluster_name_colors"] = [
"#dedede",
"#023fa5",
"#7d87b9",
"#bec1d4",
"#d6bcc0",
"#bb7784",
"#8e063b",
"#4a6fe3",
"#8595e1",
"#b5bbe3",
"#e6afb9",
"#e07b91",
"#d33f6a",
"#11c638",
]
adata
AnnData object with n_obs × n_vars = 54044 × 27998
obs: 'cluster_name', 'day', 'is_doublet'
uns: 'cluster_name_colors'
obsm: 'X_umap'
layers: 'ambiguous', 'matrix', 'spliced', 'unspliced'
Data preprocessing#
scv.pp.filter_and_normalize(adata, min_counts=20, n_top_genes=2000)
Filtered out 11060 genes that are detected 20 counts (spliced).
Normalized count data: X, spliced, unspliced.
Extracted 2000 highly variable genes.
Logarithmized X.
/vol/storage/philipp/code/scvelo_dev/scvelo/preprocessing/utils.py:705: DeprecationWarning: `log1p` is deprecated since scVelo v0.3.0 and will be removed in a future version. Please use `log1p` from `scanpy.pp` instead.
log1p(adata)
sc.pp.pca(adata)
sc.pp.neighbors(adata, random_state=0)
scv.pp.moments(adata, n_pcs=None, n_neighbors=None)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:16)
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:02:37)
computing moments based on connectivities
finished (0:00:04) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
if running_in_notebook():
scv.pl.scatter(
adata, basis="umap", c="cluster_name", title="", dpi=250, legend_fontsize=12, legend_fontweight="normal"
)

RNA velocity inference#
if (DATA_DIR / "pharyngeal_endoderm" / "results" / "adata_velo_fit-full_data.h5ad").is_file():
adata = sc.read(DATA_DIR / "pharyngeal_endoderm" / "results" / "adata_velo_fit-full_data.h5ad")
else:
scv.tl.recover_dynamics(adata, n_jobs=N_JOBS)
scv.tl.velocity(adata, mode="dynamical")
adata.write(DATA_DIR / "pharyngeal_endoderm" / "results" / "adata_velo_fit-full_data.h5ad", compression="gzip")
CellRank analysis#
Kernel#
vk = cr.kernels.VelocityKernel(adata).compute_transition_matrix()
ck = cr.kernels.ConnectivityKernel(adata).compute_transition_matrix()
combined_kernel = 0.8 * vk + 0.2 * ck
Computing transition matrix using `'deterministic'` model
Using `softmax_scale=3.4602`
Finish (0:00:29)
Computing transition matrix based on `adata.obsp['connectivities']`
DEBUG: Density normalizing the transition matrix
Finish (0:00:00)
Estimator#
estimator = cr.estimators.GPCCA(combined_kernel)
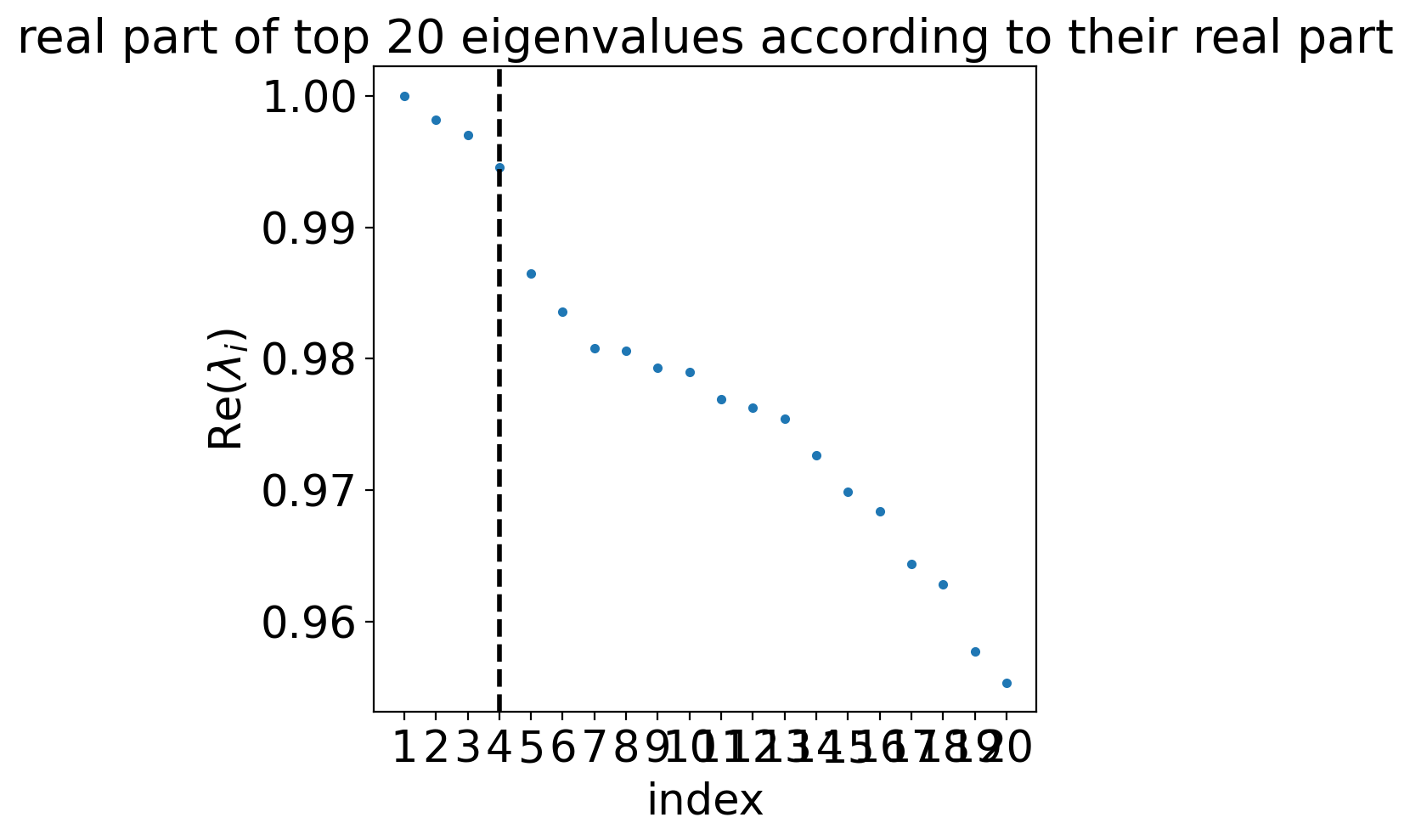
estimator.compute_schur(n_components=20)
estimator.plot_spectrum(real_only=True)
Computing Schur decomposition
Adding `adata.uns['eigendecomposition_fwd']`
`.schur_vectors`
`.schur_matrix`
`.eigendecomposition`
Finish (0:00:02)
terminal_states = [
"late_Dlx2",
"late_Runx1",
"parathyroid",
"cTEC",
"mTEC",
"late_Grhl3",
"late_Pitx2",
"ubb",
"thyroid",
"late_Dmrt1",
"late_respiratory",
]
cluster_key = "cluster_name"
if (DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-vk.csv").is_file():
tsi_df = pd.read_csv(DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-vk.csv")
estimator._tsi = AnnData(tsi_df, uns={"terminal_states": terminal_states, "cluster_key": cluster_key})
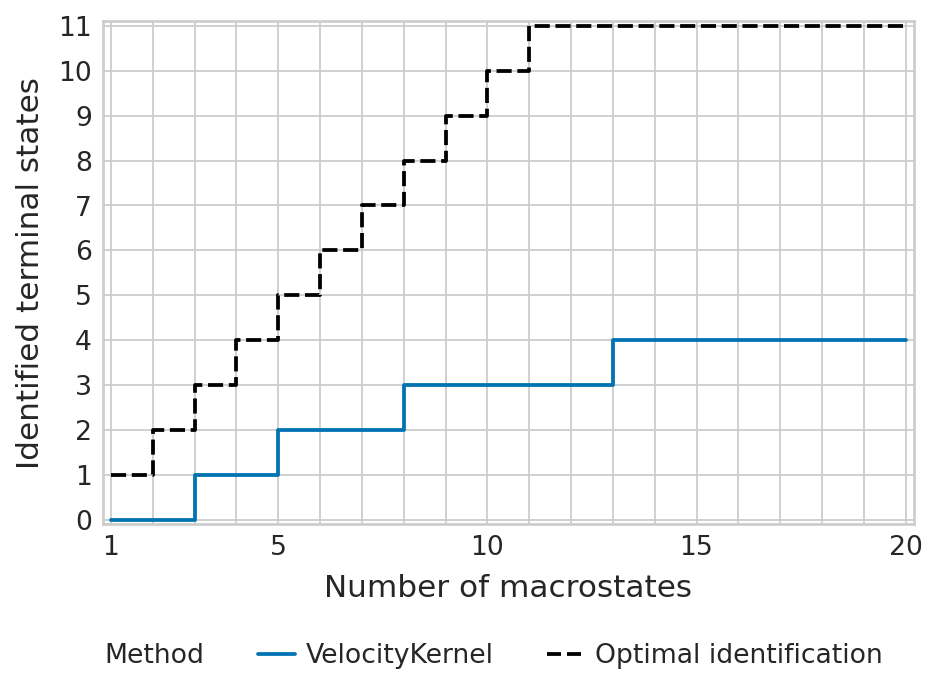
tsi_score = estimator.tsi(n_macrostates=20, terminal_states=terminal_states, cluster_key=cluster_key)
else:
tsi_score = estimator.tsi(n_macrostates=20, terminal_states=terminal_states, cluster_key=cluster_key)
estimator._tsi.to_df().to_csv(DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-vk.csv", index=False)
print(f"TSI score: {tsi_score:.2f}")
TSI score: 0.33
/vol/storage/miniconda3/envs/cr2-py38/lib/python3.8/site-packages/anndata/_core/anndata.py:121: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
# For nice name in figure legend
estimator.kernel.__class__.__name__ = "VelocityKernel"
palette = {"VelocityKernel": "#0173b2", "Optimal identification": "#000000"}
if SAVE_FIGURES:
fpath = FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"tsi-full_data-vk.{FIGURE_FORMAT}"
else:
fpath = None
with mplscience.style_context():
sns.set_style(style="whitegrid")
estimator.plot_tsi(palette=palette, save=fpath)
plt.show()