Pharyngeal endoderm development analysis with the RealTimeKernel#
Import packages#
import sys
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import mplscience
import seaborn as sns
import cellrank as cr
import scanpy as sc
import scvelo as scv
import wot
from anndata import AnnData
from cr2 import get_state_purity, plot_state_purity, running_in_notebook
sys.path.extend(["../../../", "."])
from paths import DATA_DIR, FIG_DIR # isort: skip # noqa: E402
Global seed set to 0
General settings#
sc.settings.verbosity = 2
cr.settings.verbosity = 4
scv.settings.verbosity = 3
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=20, color_map="viridis")
SAVE_FIGURES = False
if SAVE_FIGURES:
(FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm").mkdir(parents=True, exist_ok=True)
FIGURE_FORMAT = "pdf"
(DATA_DIR / "pharyngeal_endoderm" / "results").mkdir(parents=True, exist_ok=True)
Constants#
# fmt: off
S_GENES = [
"Mcm5", "Pcna", "Tyms", "Fen1", "Mcm2", "Mcm4", "Rrm1", "Ung", "Gins2",
"Mcm6", "Cdca7", "Dtl", "Prim1", "Uhrf1", "Mlf1ip", "Hells", "Rfc2",
"Rpa2", "Nasp", "Rad51ap1", "Gmnn", "Wdr76", "Slbp", "Ccne2", "Ubr7",
"Pold3", "Msh2", "Atad2", "Rad51", "Rrm2", "Cdc45", "Cdc6", "Exo1",
"Tipin", "Dscc1", "Blm", "Casp8ap2", "Usp1", "Clspn", "Pola1", "Chaf1b",
"Brip1", "E2f8",
]
G2M_GENES = [
"Hmgb2", "Cdk1", "Nusap1", "Ube2c", "Birc5", "Tpx2", "Top2a", "Ndc80",
"Cks2", "Nuf2", "Cks1b", "Mki67", "Tmpo", "Cenpf", "Tacc3", "Fam64a",
"Smc4", "Ccnb2", "Ckap2l", "Ckap2", "Aurkb", "Bub1", "Kif11", "Anp32e",
"Tubb4b", "Gtse1", "Kif20b", "Hjurp", "Cdca3", "Hn1", "Cdc20", "Ttk",
"Cdc25c", "Kif2c", "Rangap1", "Ncapd2", "Dlgap5", "Cdca2", "Cdca8",
"Ect2", "Kif23", "Hmmr", "Aurka", "Psrc1", "Anln", "Lbr", "Ckap5",
"Cenpe", "Ctcf", "Nek2", "G2e3", "Gas2l3", "Cbx5", "Cenpa",
]
# fmt: on
Data loading#
adata = sc.read(DATA_DIR / "pharyngeal_endoderm" / "raw" / "adata_pharynx.h5ad")
adata.obsm["X_umap"] = adata.obs[["UMAP1", "UMAP2"]].values
adata.obs["day"] = adata.obs["day_str"].astype(float)
adata.obs = adata.obs[["cluster_name", "day", "is_doublet"]]
adata
AnnData object with n_obs × n_vars = 54044 × 27998
obs: 'cluster_name', 'day', 'is_doublet'
obsm: 'X_umap'
Data preprocessing#
sc.pp.highly_variable_genes(adata)
extracting highly variable genes
finished (0:00:15)
sc.tl.pca(adata)
sc.pp.neighbors(adata, n_pcs=30, n_neighbors=30)
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:03)
computing neighbors
using 'X_pca' with n_pcs = 30
finished (0:02:38)
if running_in_notebook():
scv.pl.scatter(
adata, basis="umap", c="cluster_name", title="", dpi=250, legend_fontsize=12, legend_fontweight="normal"
)
if SAVE_FIGURES:
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(adata, basis="umap", c="cluster_name", legend_loc=False, title="", ax=ax)
fig.savefig(
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"umap_colored_by_cell_type_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(adata, basis="umap", c="day", legend_loc=False, title="", ax=ax)
fig.savefig(
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"umap_colored_by_day_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)

CellRank#
Kernel#
if not (DATA_DIR / "pharyngeal_endoderm" / "tmaps_full_data").exists():
ot_model = wot.ot.OTModel(adata)
ot_model.compute_all_transport_maps(tmap_out=DATA_DIR / "pharyngeal_endoderm" / "tmaps_full_data" / "tmaps")
adata.obs["day"] = adata.obs["day"].astype("category")
rtk = cr.kernels.RealTimeKernel.from_wot(
adata, path=DATA_DIR / "pharyngeal_endoderm" / "tmaps_full_data", time_key="day"
)
rtk.compute_transition_matrix(
growth_iters=3, growth_rate_key="growth_rate_init", self_transitions="all", conn_weight=0.1
)
WARNING: Your filename has more than two extensions: ['.5_11', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
WARNING: Your filename has more than two extensions: ['.5_11', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
WARNING: Your filename has more than two extensions: ['.5_12', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
WARNING: Your filename has more than two extensions: ['.5_12', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
WARNING: Your filename has more than two extensions: ['.5_10', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
WARNING: Your filename has more than two extensions: ['.5_10', '.5', '.h5ad'].
Only considering the two last: ['.5', '.h5ad'].
Using automatic `threshold=0.0022490203846246004`
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:00:02)
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:00:02)
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:00:02)
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:00:01)
RealTimeKernel[n=54044, growth_iters=3, growth_rate_key='growth_rate_init', threshold='auto', self_transitions='all']
Estimator#
estimator = cr.estimators.GPCCA(rtk)
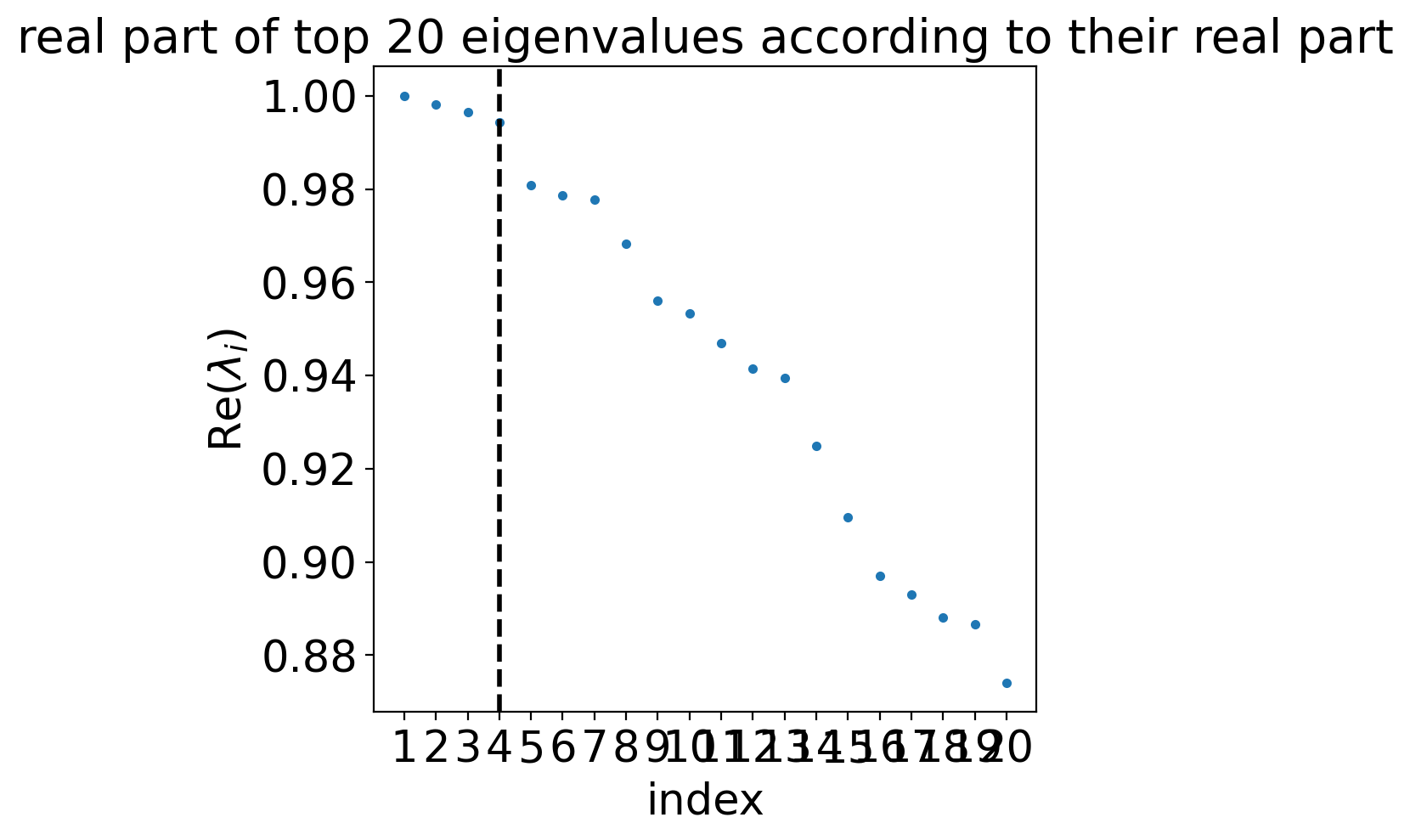
estimator.compute_schur(n_components=20)
estimator.plot_spectrum(real_only=True)
plt.show()
Computing Schur decomposition
Adding `adata.uns['eigendecomposition_fwd']`
`.schur_vectors`
`.schur_matrix`
`.eigendecomposition`
Finish (0:00:04)
terminal_states = [
"late_Dlx2",
"late_Runx1",
"parathyroid",
"cTEC",
"mTEC",
"late_Grhl3",
"late_Pitx2",
"ubb",
"thyroid",
"late_Dmrt1",
"late_respiratory",
]
cluster_key = "cluster_name"
if (DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-rtk.csv").is_file():
tsi_df = pd.read_csv(DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-rtk.csv")
estimator._tsi = AnnData(tsi_df, uns={"terminal_states": terminal_states, "cluster_key": cluster_key})
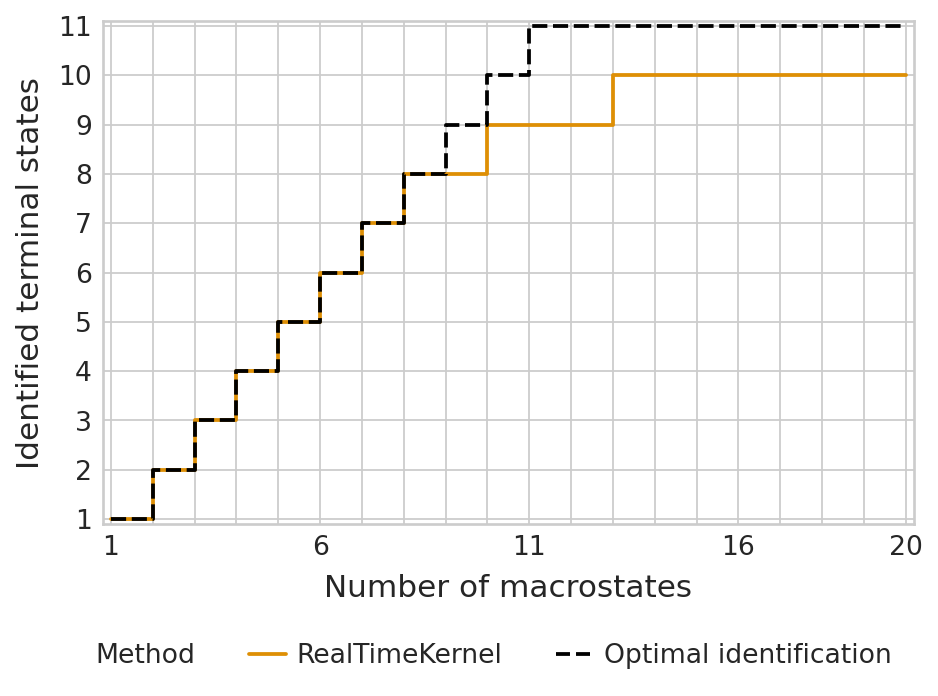
tsi_score = estimator.tsi(n_macrostates=20, terminal_states=terminal_states, cluster_key=cluster_key)
else:
tsi_score = estimator.tsi(n_macrostates=20, terminal_states=terminal_states, cluster_key=cluster_key)
estimator._tsi.to_df().to_csv(DATA_DIR / "pharyngeal_endoderm" / "results" / "tsi-full_data-rtk.csv", index=False)
print(f"TSI score: {tsi_score:.2f}")
TSI score: 0.92
/vol/storage/miniconda3/envs/cr2-py38/lib/python3.8/site-packages/anndata/_core/anndata.py:121: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
palette = {"RealTimeKernel": "#DE8F05", "Optimal identification": "#000000"}
if SAVE_FIGURES:
fpath = FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"tsi-full_data-rtk.{FIGURE_FORMAT}"
else:
fpath = None
with mplscience.style_context():
sns.set_style(style="whitegrid")
estimator.plot_tsi(palette=palette, save=fpath)
plt.show()
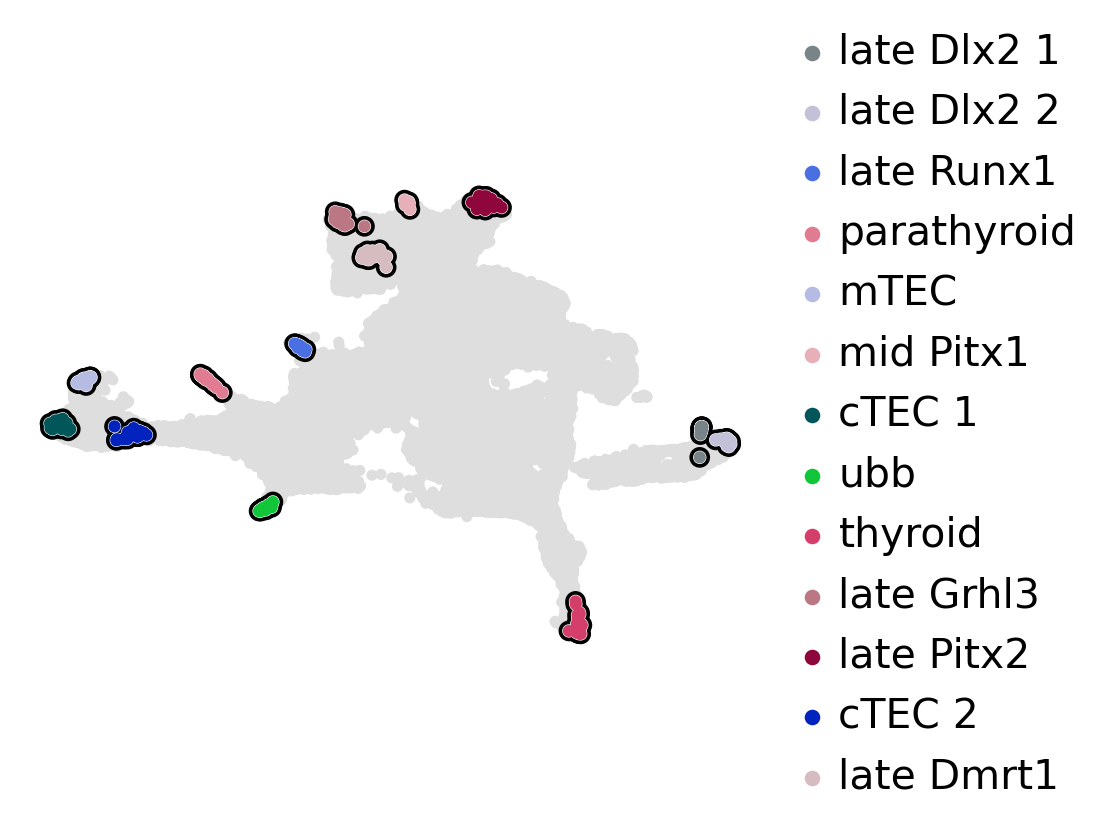
estimator.compute_macrostates(n_states=13, cluster_key="cluster_name")
if running_in_notebook():
estimator.plot_macrostates(which="all", basis="umap", title="", legend_loc="right", size=100)
if SAVE_FIGURES:
fpath = (
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"umap_colored_by_macrostates_full_data.{FIGURE_FORMAT}"
)
estimator.plot_macrostates(which="all", basis="umap", title="", legend_loc=False, size=100, save=fpath)
Computing `13` macrostates
DEBUG: Setting the macrostates using macrostates memberships
DEBUG: Raising an exception if there are less than `6` cells.
Adding `.macrostates`
`.macrostates_memberships`
`.coarse_T`
`.coarse_initial_distribution
`.coarse_stationary_distribution`
`.schur_vectors`
`.schur_matrix`
`.eigendecomposition`
Finish (0:01:59)
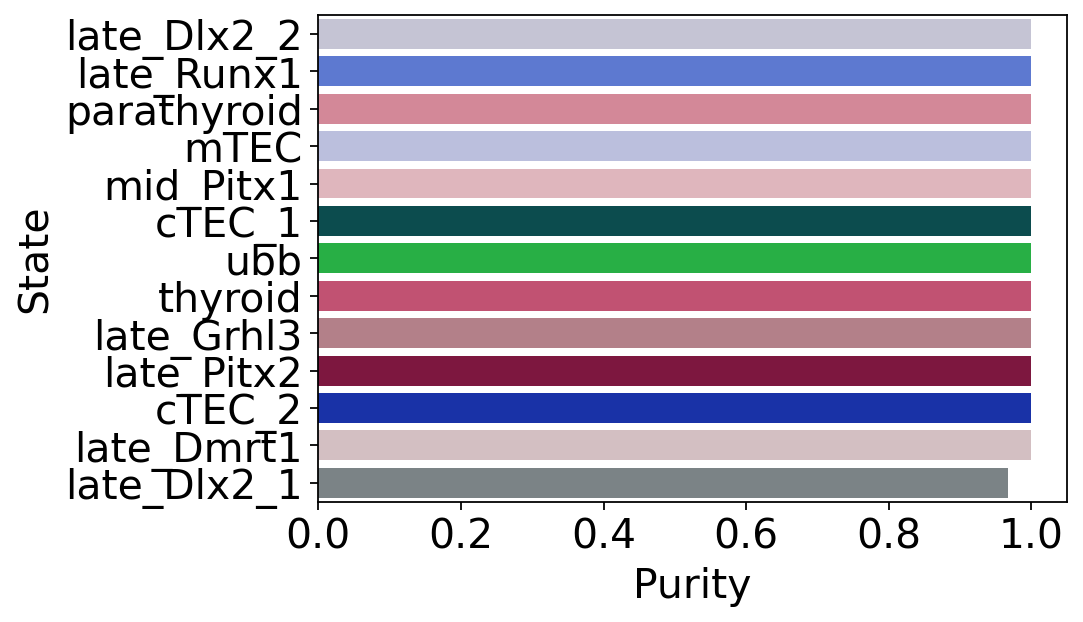
macrostate_purity = get_state_purity(adata, estimator, states="macrostates", obs_col="cluster_name")
print(f"Mean purity: {np.mean(list(macrostate_purity.values()))}")
if running_in_notebook():
if SAVE_FIGURES:
fpath = FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"macrostate_purity_full_data.{FIGURE_FORMAT}"
else:
fpath = None
palette = dict(zip(estimator.macrostates.cat.categories, estimator._macrostates.colors))
plot_state_purity(macrostate_purity, palette=palette, fpath=fpath, format=FIGURE_FORMAT)
plt.show()
Mean purity: 0.9974358974358974
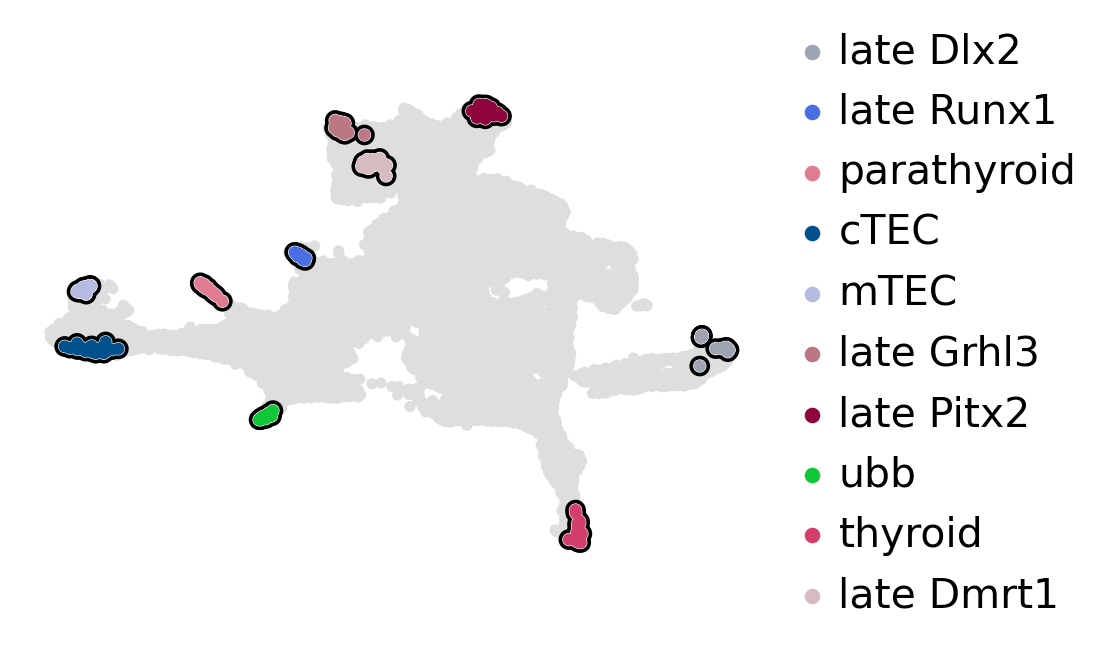
estimator.set_terminal_states(
[
"late_Dlx2_1, late_Dlx2_2",
"late_Runx1",
"parathyroid",
"cTEC_1, cTEC_2",
"mTEC",
"late_Grhl3",
"late_Pitx2",
"ubb",
"thyroid",
"late_Dmrt1",
]
)
estimator.rename_terminal_states({"cTEC_1, cTEC_2": "cTEC", "late_Dlx2_1, late_Dlx2_2": "late_Dlx2"})
if running_in_notebook():
estimator.plot_macrostates(which="terminal", basis="umap", title="", legend_loc="right", size=100)
if SAVE_FIGURES:
fpath = (
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"umap_colored_by_terminal_states_full_data.{FIGURE_FORMAT}"
)
estimator.plot_macrostates(which="terminal", basis="umap", title="", legend_loc=False, size=100, save=fpath)
DEBUG: Raising an exception if there are less than `6` cells.
Adding `adata.obs['term_states_fwd']`
`adata.obs['term_states_fwd_probs']`
`.terminal_states`
`.terminal_states_probabilities`
`.terminal_states_memberships
Finish`
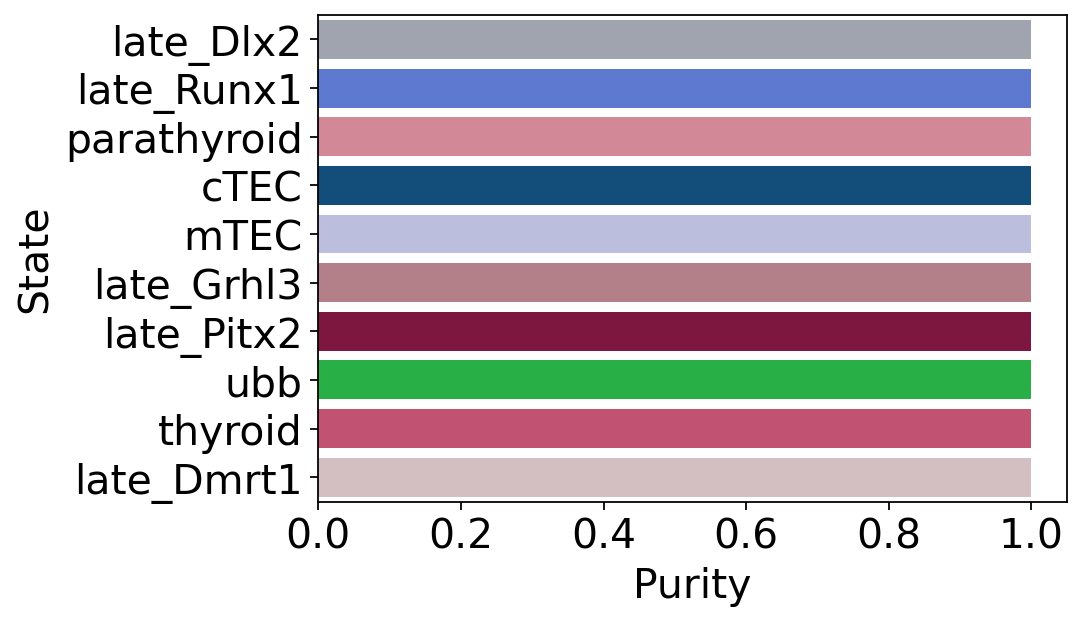
terminal_state_purity = get_state_purity(adata, estimator, states="terminal_states", obs_col="cluster_name")
print(f"Mean purity: {np.mean(list(terminal_state_purity.values()))}")
if running_in_notebook():
if SAVE_FIGURES:
fpath = (
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"terminal_states_purity_full_data.{FIGURE_FORMAT}"
)
else:
fpath = None
palette = dict(zip(estimator.terminal_states.cat.categories, estimator._term_states.colors))
plot_state_purity(terminal_state_purity, palette=palette, fpath=fpath, format=FIGURE_FORMAT)
plt.show()
Mean purity: 1.0
estimator.compute_fate_probabilities(solver="gmres", use_petsc=True)
Computing fate probabilities
DEBUG: Solving the linear system using `PETSc` solver `'gmres'` on `1` core(s) with no preconditioner and `tol=1e-06`
Adding `adata.obsm['lineages_fwd']`
`.fate_probabilities`
Finish (0:00:03)
[0]PETSC ERROR: ------------------------------------------------------------------------
[0]PETSC ERROR: Caught signal number 13 Broken Pipe: Likely while reading or writing to a socket
[0]PETSC ERROR: Try option -start_in_debugger or -on_error_attach_debugger
[0]PETSC ERROR: or see https://petsc.org/release/faq/#valgrind and https://petsc.org/release/faq/
[0]PETSC ERROR: configure using --with-debugging=yes, recompile, link, and run
[0]PETSC ERROR: to get more information on the crash.
Abort(59) on node 0 (rank 0 in comm 0): application called MPI_Abort(MPI_COMM_WORLD, 59) - process 0
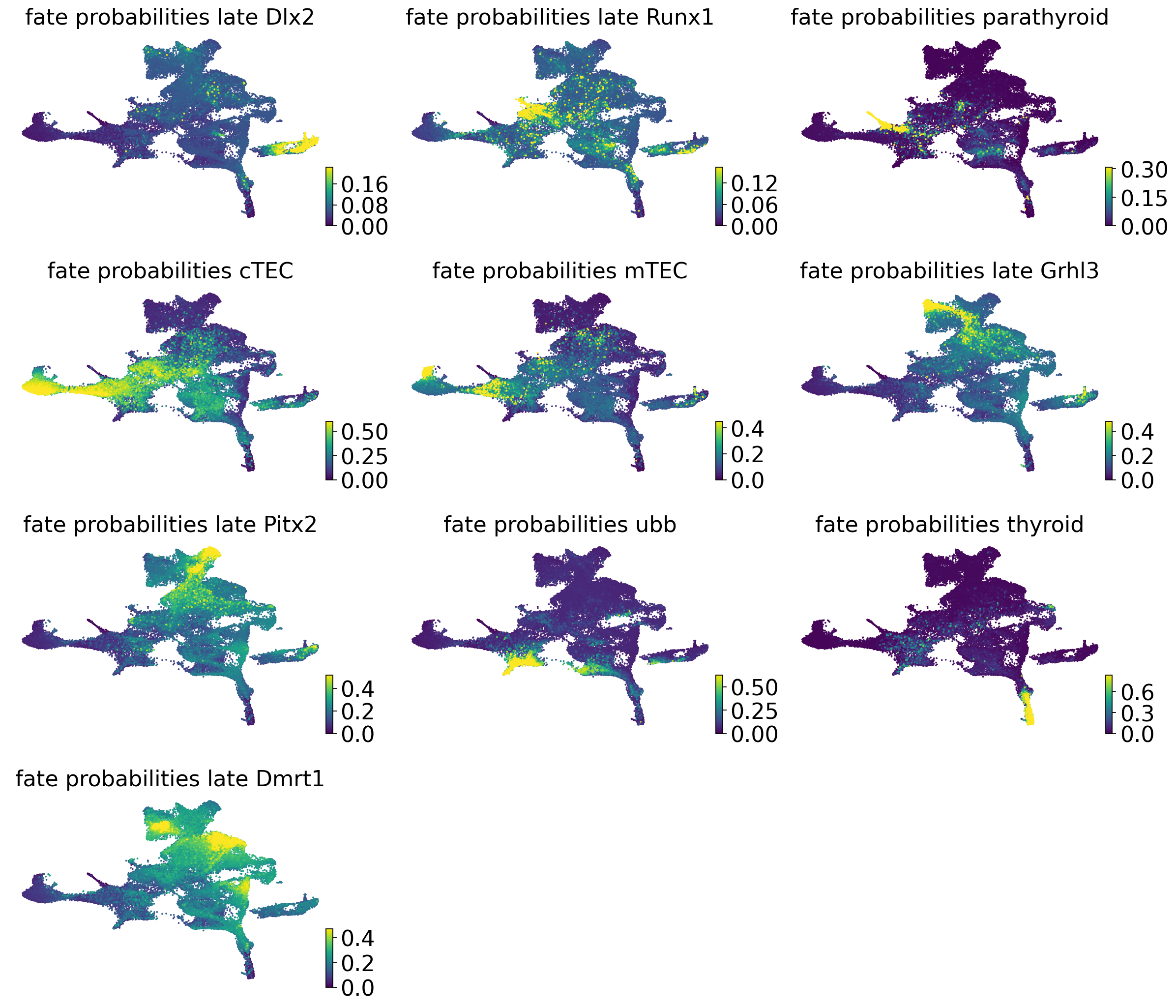
estimator.compute_fate_probabilities()
if running_in_notebook():
estimator.plot_fate_probabilities(same_plot=False, basis="umap", perc=[0, 99], ncols=3)
if SAVE_FIGURES:
for terminal_state in estimator.terminal_states.cat.categories:
adata.obs[f"fate_prob_{terminal_state}"] = adata.obsm["lineages_fwd"][:, terminal_state].X.squeeze()
fig, ax = plt.subplots(figsize=(6, 4))
if running_in_notebook():
scv.pl.scatter(
adata,
basis="umap",
color=f"fate_prob_{terminal_state}",
cmap="viridis",
title="",
colorbar=False,
ax=ax,
)
fig.savefig(
FIG_DIR
/ "realtime_kernel"
/ "pharyngeal_endoderm"
/ f"fate_prob_{terminal_state}_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
Computing fate probabilities
DEBUG: Solving the linear system using `PETSc` solver `'gmres'` on `1` core(s) with no preconditioner and `tol=1e-06`
Adding `adata.obsm['lineages_fwd']`
`.fate_probabilities`
Finish (0:00:03)
[0]PETSC ERROR: ------------------------------------------------------------------------
[0]PETSC ERROR: Caught signal number 13 Broken Pipe: Likely while reading or writing to a socket
[0]PETSC ERROR: Try option -start_in_debugger or -on_error_attach_debugger
[0]PETSC ERROR: or see https://petsc.org/release/faq/#valgrind and https://petsc.org/release/faq/
[0]PETSC ERROR: configure using --with-debugging=yes, recompile, link, and run
[0]PETSC ERROR:
Driver analysis#
adata.obs["cluster_name_"] = adata.obs["cluster_name"].copy().astype(str).astype("category")
drivers_ctec = estimator.compute_lineage_drivers(
return_drivers=True, cluster_key="cluster_name_", lineages=["cTEC"], clusters=["nan", "early_thymus", "cTEC"]
)
gene_names = drivers_ctec.loc[
~(
drivers_ctec.index.str.startswith(("mt.", "Rpl", "Rps", "^Hb[^(p)]"))
| drivers_ctec.index.isin(S_GENES + G2M_GENES)
),
:,
].index
if running_in_notebook():
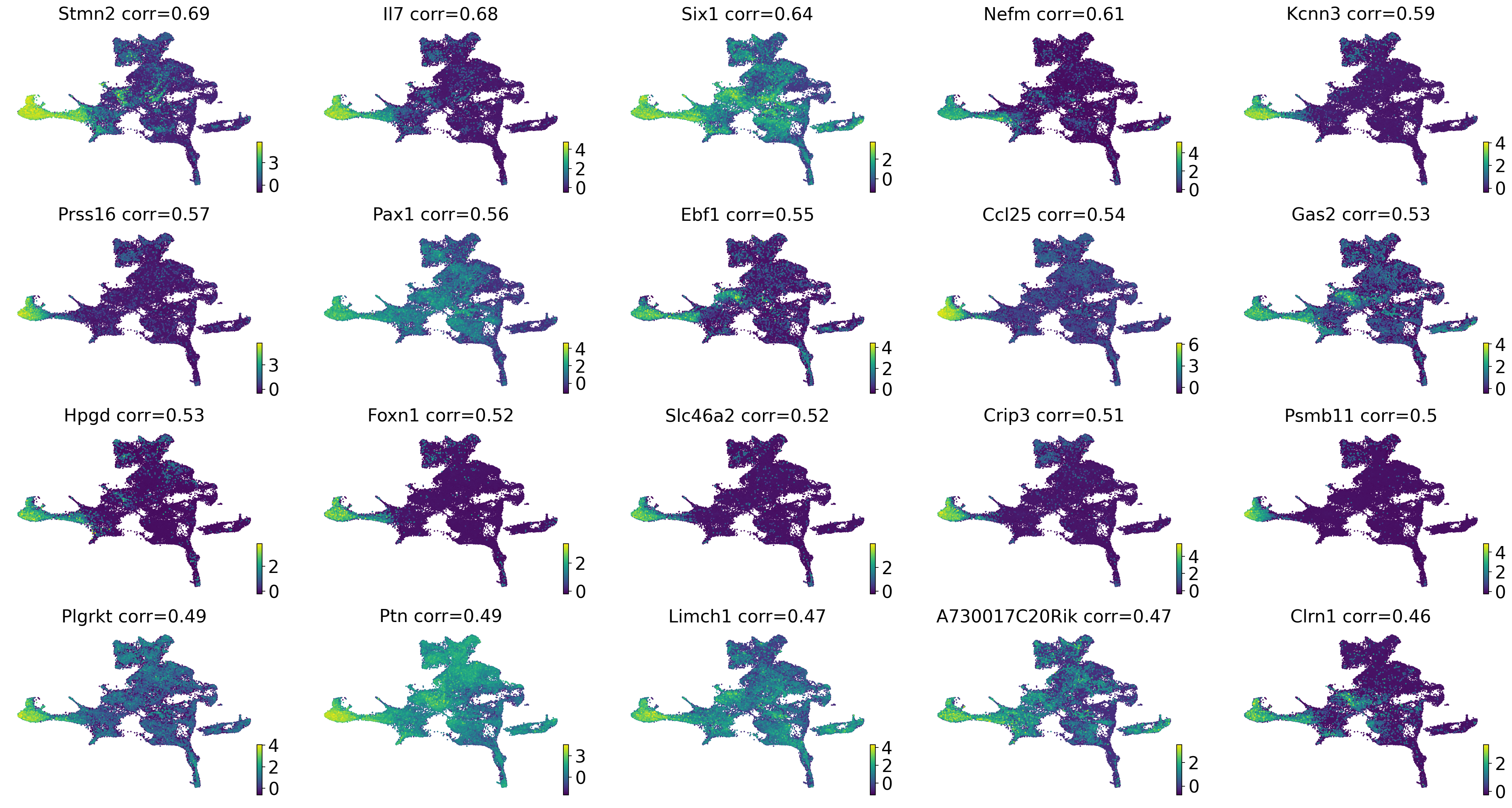
estimator.plot_lineage_drivers(lineage="cTEC", n_genes=20, ncols=5, title_fmt="{gene} corr={corr:.2}")
if SAVE_FIGURES:
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(
adata, basis="umap", c="Foxn1", cmap="viridis", title="", legend_loc=None, colorbar=False, ax=ax, s=25
)
fig.savefig(
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"umap_colored_by_foxn1_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
np.where(gene_names == "Foxn1")
DEBUG: Computing correlations for lineages `['cTEC']` restricted to clusters `['nan', 'early_thymus', 'cTEC']` in layer `X` with `use_raw=False`
Adding `adata.varm['terminal_lineage_drivers']`
`.lineage_drivers`
Finish (0:00:09)
(array([11]),)
drivers_parathyroid = estimator.compute_lineage_drivers(
return_drivers=True, cluster_key="cluster_name_", lineages=["parathyroid"], clusters=["nan"]
)
gene_names = drivers_parathyroid.loc[
~(
drivers_parathyroid.index.str.startswith(("mt.", "Rpl", "Rps", "^Hb[^(p)]"))
| drivers_parathyroid.index.isin(S_GENES + G2M_GENES)
),
:,
].index
if running_in_notebook():
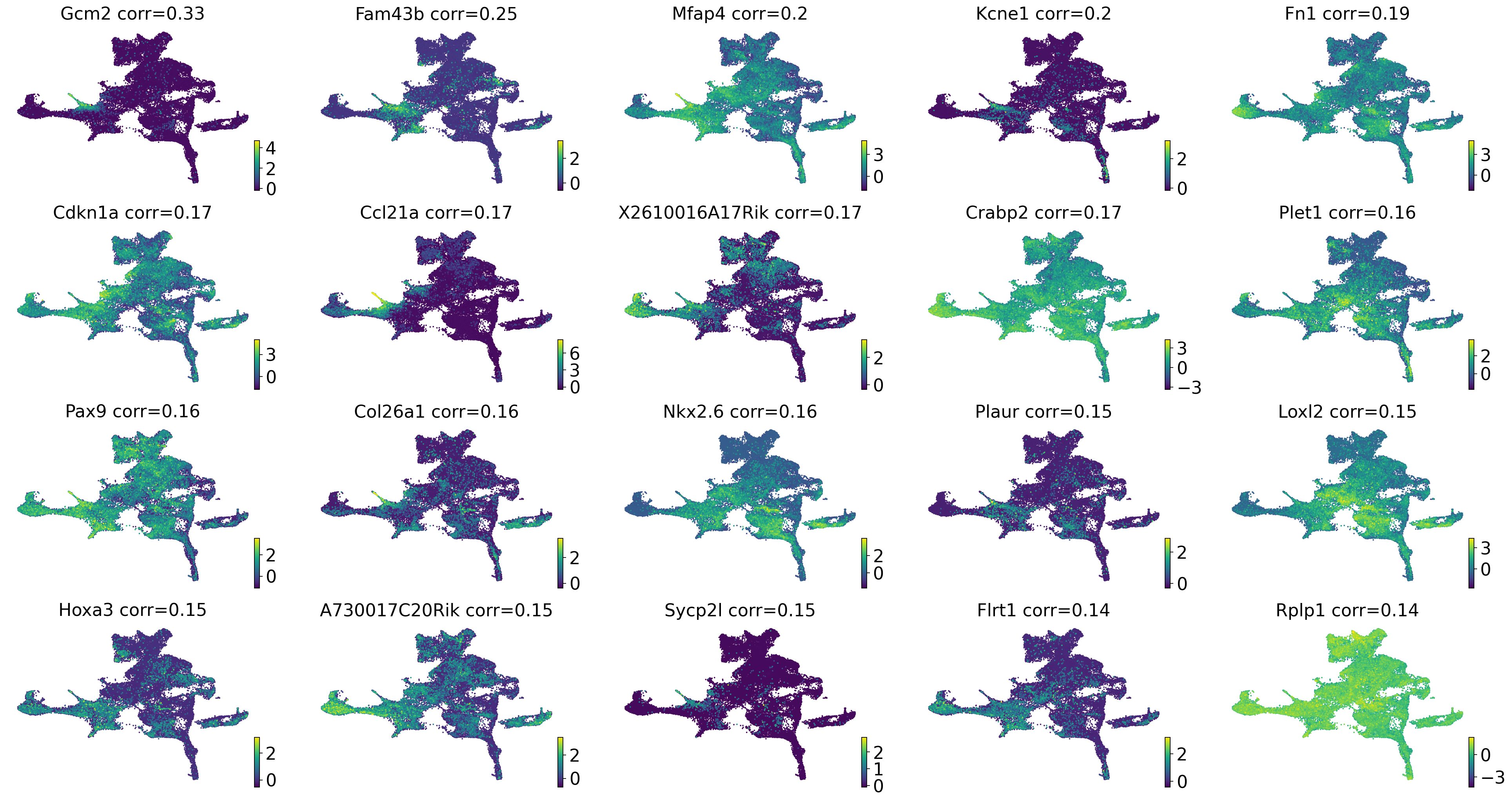
estimator.plot_lineage_drivers(lineage="parathyroid", n_genes=20, ncols=5, title_fmt="{gene} corr={corr:.2}")
if SAVE_FIGURES:
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(
adata, basis="umap", c="Gcm2", cmap="viridis", title="", legend_loc=None, colorbar=False, ax=ax, s=25
)
fig.savefig(
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"umap_colored_by_gcm2_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
np.where(gene_names == "Gcm2")
DEBUG: Computing correlations for lineages `['parathyroid']` restricted to clusters `['nan']` in layer `X` with `use_raw=False`
Adding `adata.varm['terminal_lineage_drivers']`
`.lineage_drivers`
Finish (0:00:07)
(array([0]),)
drivers_thyroid = estimator.compute_lineage_drivers(
return_drivers=True, cluster_key="cluster_name_", lineages=["thyroid"], clusters=["nan"]
)
gene_names = drivers_thyroid.loc[
~(
drivers_thyroid.index.str.startswith(("mt.", "Rpl", "Rps", "^Hb[^(p)]"))
| drivers_thyroid.index.isin(S_GENES + G2M_GENES)
),
:,
].index
if running_in_notebook():
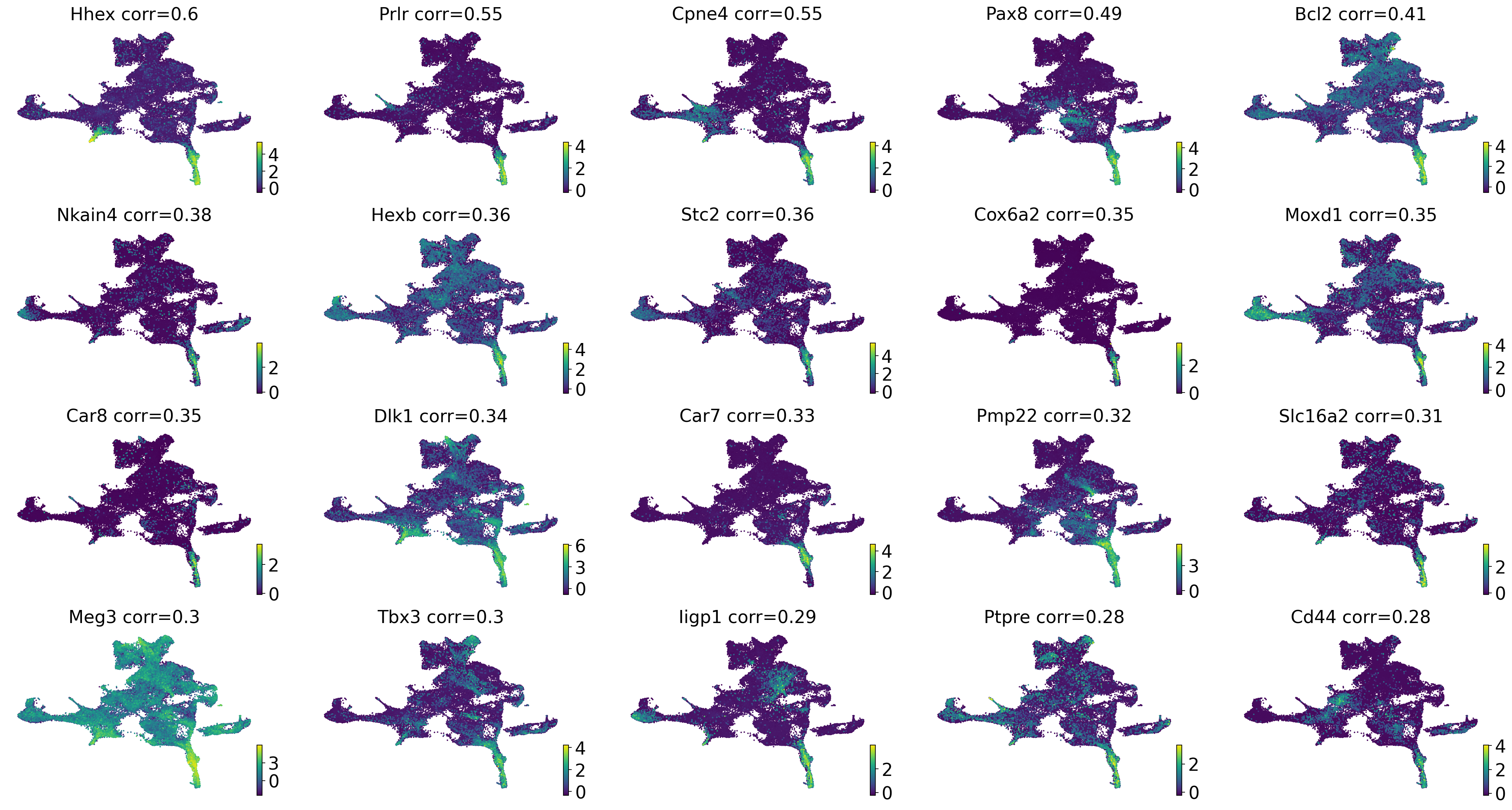
estimator.plot_lineage_drivers(lineage="thyroid", n_genes=20, ncols=5, title_fmt="{gene} corr={corr:.2}")
if SAVE_FIGURES:
fig, ax = plt.subplots(figsize=(6, 4))
scv.pl.scatter(
adata, basis="umap", c="Hhex", cmap="viridis", title="", legend_loc=None, colorbar=False, ax=ax, s=25
)
fig.savefig(
FIG_DIR / "realtime_kernel" / "pharyngeal_endoderm" / f"umap_colored_by_hhex_full_data.{FIGURE_FORMAT}",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
np.where(gene_names == "Hhex")
DEBUG: Computing correlations for lineages `['thyroid']` restricted to clusters `['nan']` in layer `X` with `use_raw=False`
Adding `adata.varm['terminal_lineage_drivers']`
`.lineage_drivers`
Finish (0:00:07)
(array([0]),)