Driver ranking benchmark#
Benchmark driver ranking.
Library import#
import pandas as pd
import scipy
from sklearn.metrics import roc_auc_score
import matplotlib.pyplot as plt
import mplscience
import seaborn as sns
import cellrank as cr
import scanpy as sc
import scvelo as scv
from rgv_tools import DATA_DIR, FIG_DIR
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
warnings.warn(msg, FutureWarning)
General settings#
plt.rcParams["svg.fonttype"] = "none"
sns.reset_defaults()
sns.reset_orig()
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=14, color_map="viridis")
Constants#
DATASET = "zebrafish"
SAVE_FIGURES = False
if SAVE_FIGURES:
(FIG_DIR / DATASET).mkdir(parents=True, exist_ok=True)
TERMINAL_STATES = [
"mNC_head_mesenchymal",
"mNC_arch2",
"mNC_hox34",
"Pigment",
]
Data loading#
adata = sc.read_h5ad(DATA_DIR / DATASET / "processed" / "adata_run_regvelo.h5ad")
Driver ranking analysis#
Correlating gene expression with cell fate probabilities#
vk = cr.kernels.VelocityKernel(adata)
vk.compute_transition_matrix()
ck = cr.kernels.ConnectivityKernel(adata).compute_transition_matrix()
kernel = 0.8 * vk + 0.2 * ck
100%|██████████| 697/697 [00:31<00:00, 22.18cell/s]
100%|██████████| 697/697 [00:00<00:00, 2299.24cell/s]
estimator = cr.estimators.GPCCA(kernel)
## evaluate the fate prob on original space
estimator.compute_macrostates(n_states=8, cluster_key="cell_type")
estimator.set_terminal_states(TERMINAL_STATES)
estimator.compute_fate_probabilities()
WARNING: Unable to import `petsc4py` or `slepc4py`. Using `method='brandts'`
WARNING: For `method='brandts'`, dense matrix is required. Densifying
WARNING: Unable to import petsc4py. For installation, please refer to: https://petsc4py.readthedocs.io/en/stable/install.html.
Defaulting to `'gmres'` solver.
100%|██████████| 4/4 [00:00<00:00, 32.14/s]
df = estimator.compute_lineage_drivers(cluster_key="cell_type")
fate_prob = pd.DataFrame(
estimator.adata.obsm["lineages_fwd"], columns=estimator.adata.obsm["lineages_fwd"].names.tolist()
)
Correlating gene activity with cell fate probabilities#
GeneActivity = pd.read_csv(DATA_DIR / DATASET / "raw" / "GeneActivity.csv", index_col=0)
gene_symbol = [i.replace("_pos", "").replace("_neg", "") for i in GeneActivity.index.tolist()]
ind = [i in adata.var_names[adata.var["TF"]] for i in gene_symbol]
GeneActivity = GeneActivity.iloc[ind, :]
gene_symbol = [i.replace("_pos", "").replace("_neg", "") for i in GeneActivity.index.tolist()]
df_a = pd.DataFrame(columns=df.columns.tolist())
for i in fate_prob.columns.tolist():
cor = []
pval = []
for g in GeneActivity.index.tolist():
res = scipy.stats.pearsonr(GeneActivity.loc[g, :], fate_prob.loc[:, i])
cor.append(res[0])
pval.append(res[1])
df_a[i + "_corr"] = cor
df_a[i + "_pval"] = pval
df_a.index = GeneActivity.index.tolist()
Predict known drivers#
driver_head_mes = ["nr2f5", "nr2f2", "sox9b", "twist1a", "twist1b"]
driver_pigment = ["sox10", "mitfa", "tfec", "bhlhe40", "tfap2b", "tfap2a"]
mNC head mesenchymal#
score_head_mes = []
label_ga = [1 if i in driver_head_mes else 0 for i in df_a.index.tolist()]
label_gex = [1 if i in driver_head_mes else 0 for i in df.index.tolist()]
for nrun in range(3):
coef_name = "coef_" + str(nrun)
coef = pd.read_csv(DATA_DIR / DATASET / "results" / coef_name, index_col=0)
label_rgv = [1 if i in driver_head_mes else 0 for i in coef.index.tolist()]
score_head_mes.append(roc_auc_score(label_rgv, coef["mNC_head_mesenchymal"]))
score_head_mes.append(roc_auc_score(label_ga, df_a["mNC_head_mesenchymal_corr"]))
score_head_mes.append(roc_auc_score(label_gex, df["mNC_head_mesenchymal_corr"]))
Pigment#
score_pigment = []
for nrun in range(3):
coef_name = "coef_" + str(nrun)
coef = pd.read_csv(DATA_DIR / DATASET / "results" / coef_name, index_col=0)
label_rgv = [1 if i in driver_pigment else 0 for i in coef.index.tolist()]
score_pigment.append(roc_auc_score(label_rgv, coef["Pigment"]))
label_ga = [1 if i in driver_pigment else 0 for i in df_a.index.tolist()]
label_gex = [1 if i in driver_pigment else 0 for i in df.index.tolist()]
score_pigment.append(roc_auc_score(label_ga, df_a["Pigment_corr"]))
score_pigment.append(roc_auc_score(label_gex, df["Pigment_corr"]))
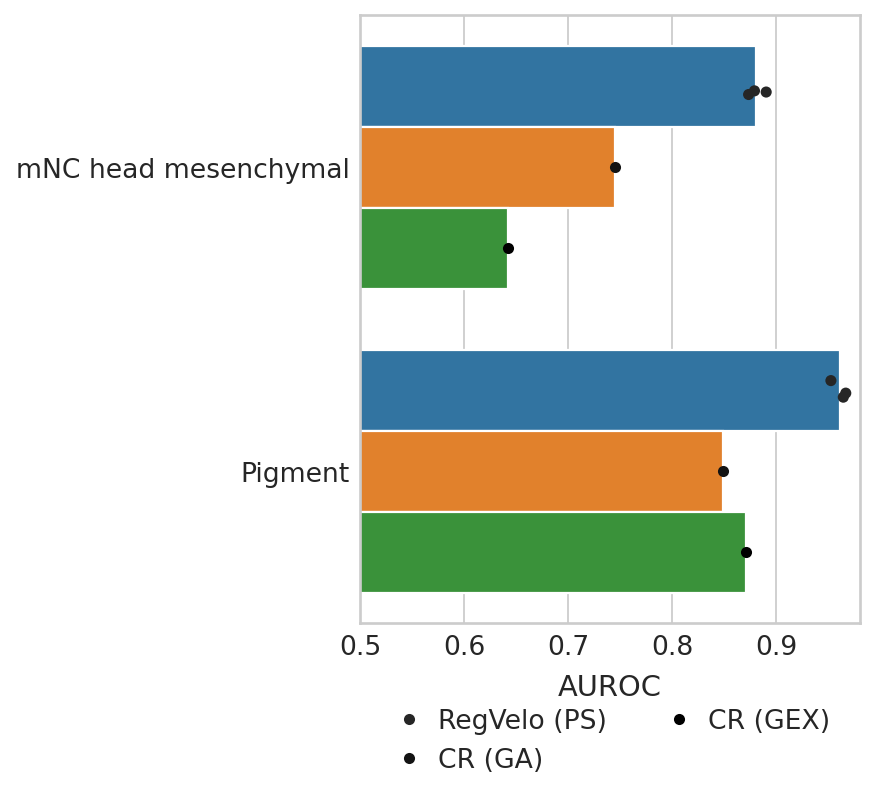
Plot results#
dat = pd.DataFrame({"AUROC": score_head_mes, "method": ["RegVelo (PS)"] * 3 + ["CR (GA)"] + ["CR (GEX)"]})
dat2 = pd.DataFrame({"AUROC": score_pigment, "method": ["RegVelo (PS)"] * 3 + ["CR (GA)"] + ["CR (GEX)"]})
dat["Lineage"] = "mNC head mesenchymal"
dat2["Lineage"] = "Pigment"
df = pd.concat([dat, dat2], axis=0)
with mplscience.style_context():
sns.set_style(style="whitegrid")
fig, ax = plt.subplots(figsize=(4, 5))
# Plot the barplot without error bars
sns.barplot(data=df, y="Lineage", x="AUROC", hue="method", ax=ax, ci=None)
# Add jittered dots
sns.stripplot(data=df, y="Lineage", x="AUROC", hue="method", dodge=True, color="black", ax=ax, jitter=True)
# Remove the duplicate legend
handles, labels = ax.get_legend_handles_labels()
ax.legend(handles[3:6], labels[3:6], bbox_to_anchor=(0.5, -0.1), loc="upper center", ncol=2)
# Customize labels and other settings
ax.set(ylabel="", xlabel="AUROC")
ax.set_xlabel(xlabel="AUROC", fontsize=13)
ax.set_xlim(0.5, 0.98)
if SAVE_FIGURES:
plt.savefig(FIG_DIR / DATASET / "AUROC_ranking.svg", format="svg", transparent=True, bbox_inches="tight")
plt.show()