Split mouse neural crest data into different scale#
Library imports#
import scvelo as scv
import scanpy as sc
import numpy as np
import anndata as ad
import pandas as pd
import matplotlib.pyplot as plt
import mplscience
from collections import Counter
import pandas as pd
from rgv_tools import DATA_DIR, FIG_DIR
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo_test/lib/python3.10/site-packages/anndata/utils.py:429: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
warnings.warn(msg, FutureWarning)
General settings#
sc.settings.verbosity = 2
scv.settings.verbosity = 3
plt.rcParams["svg.fonttype"] = "none"
scv.settings.set_figure_params("scvelo", dpi=100, transparent=True, fontsize=14, color_map="viridis")
Constants#
DATASET = "mouse_neural_crest"
SAVE_DATA = True
if SAVE_DATA:
(DATA_DIR / DATASET / "processed").mkdir(parents=True, exist_ok=True)
SAVE_FIGURES = True
if SAVE_FIGURES:
(FIG_DIR / DATASET).mkdir(parents=True, exist_ok=True)
FIGURE_FORMAT = "svg"
TERMINAL_STATE = [
"Melanocytes",
"enFib",
"SC",
"Mesenchyme",
"Sensory",
"ChC",
"SatGlia",
"Gut_glia",
"Gut_neuron",
"Symp",
"BCC",
]
Location = ["Cranial", "Trunk", "DRG", "Limb", "Enteric", "Sympathoadrenal system", "Incisor"]
Functions definations#
def count_cell_types(cell_type_list, class_list):
counter = Counter(cell_type_list)
result = {cls: counter.get(cls, 0) for cls in class_list}
df = pd.DataFrame({"Cell Type": list(result.keys()), "Count": list(result.values())})
return df
Data loading#
ldata = ad.io.read_h5ad(DATA_DIR / DATASET / "raw" / "GSE201257_adata_velo_raw.h5ad")
adata = ad.io.read_h5ad(DATA_DIR / DATASET / "raw" / "GSE201257_adata_processed.h5ad")
Annotate all datasets#
We followed the annotation procedure provided by original authors, please check LouisFaure/glialfates_paper
adata.layers["GEX"] = adata.X.copy()
adata.X = sc.pp.scale(adata.layers["palantir_imp"], max_value=10)
... as `zero_center=True`, sparse input is densified and may lead to large memory consumption
sc.tl.score_genes(
adata,
["Ret", "Phox2a", "Chrna3", "Sox11"],
score_name="Gut_neuron",
)
adata.obs.Gut_neuron = adata.obs.Gut_neuron > 1.5
sc.tl.score_genes(adata, ["Prdm12", "Isl2", "Pou4f1", "Six1"], score_name="Sensory")
adata.obs.Sensory = adata.obs.Sensory > 1
sc.tl.score_genes(adata, ["Cartpt", "Prph", "Mapt", "Maoa"], score_name="Symp")
adata.obs.Symp = adata.obs.Symp > 1.5
sc.tl.score_genes(adata, ["Lum", "Dcn", "Fbn1"], score_name="enFib")
adata.obs.enFib = adata.obs.enFib > 2.5
sc.tl.score_genes(adata, ["Th", "Dbh", "Chga", "Chgb", "Slc18a1", "Slc18a2"], score_name="ChC")
adata.obs.ChC = adata.obs.ChC > 2
sc.tl.score_genes(adata, ["Phox2b", "Ctgf", "Nfia", "Tgfb2", "S100b"], score_name="Gut_glia")
adata.obs.Gut_glia = adata.obs.Gut_glia > 2.5
sc.tl.score_genes(adata, ["Sox9", "Wnt1", "Ets1", "Crabp2"], score_name="NCC")
adata.obs.NCC = adata.obs.NCC > 0.6
sc.tl.score_genes(adata, ["Prrx1", "Prrx2", "Pdgfra"], score_name="Mesenchyme")
adata.obs.Mesenchyme = adata.obs.Mesenchyme > 0.4
sc.tl.score_genes(adata, ["Dct", "Mitf", "Pmel", "Tyr"], score_name="Melanocytes")
adata.obs.Melanocytes = adata.obs.Melanocytes > 1
sc.tl.score_genes(adata, ["Fabp7", "Ptn", "Rgcc"], score_name="SatGlia")
adata.obs.SatGlia = adata.obs.SatGlia > 2.6
computing score 'Gut_neuron'
finished (0:00:01)
computing score 'Sensory'
finished (0:00:00)
computing score 'Symp'
finished (0:00:00)
computing score 'enFib'
finished (0:00:00)
computing score 'ChC'
finished (0:00:00)
computing score 'Gut_glia'
finished (0:00:00)
computing score 'NCC'
finished (0:00:01)
computing score 'Mesenchyme'
finished (0:00:00)
computing score 'Melanocytes'
finished (0:00:00)
computing score 'SatGlia'
finished (0:00:00)
sc.tl.score_genes(adata, ["Mpz", "Plp1", "Fbxo7", "Gjc3", "Pmp22", "Dhh", "Mal"], score_name="SC")
adata.obs.SC = adata.obs.SC > 1.35
computing score 'SC'
finished (0:00:00)
sc.tl.score_genes(adata, ["Prss56", "Egr2", "Wif1", "Hey2"], score_name="BCC")
adata.obs.BCC = adata.obs.BCC > 6
computing score 'BCC'
finished (0:00:00)
celltypes = [
"NCC",
"Symp",
"ChC",
"Sensory",
"Gut_glia",
"Gut_neuron",
"Melanocytes",
"SC",
"enFib",
"Mesenchyme",
"SatGlia",
"BCC",
]
adata.obs["conflict"] = adata.obs[celltypes].sum(axis=1) > 1
adata.obs.loc[adata.obs[celltypes].sum(axis=1) > 1, celltypes] = False
adata.obs.loc[adata.obs[celltypes].sum(axis=1) == 2, celltypes] = False
annot = adata.obs.loc[:, celltypes].apply(lambda x: np.argwhere(x.values), axis=1)
annot = annot[annot.apply(len) == 1]
annot = annot.apply(lambda x: np.array(celltypes)[x][0][0])
adata.obs["assignments"] = "none"
adata.obs.loc[annot.index, "assignments"] = annot.values
adata.obs["assignments"] = adata.obs["assignments"].astype("category")
adata.obs.assignments.cat.categories
Index(['BCC', 'ChC', 'Gut_glia', 'Gut_neuron', 'Melanocytes', 'Mesenchyme',
'NCC', 'SC', 'SatGlia', 'Sensory', 'Symp', 'enFib', 'none'],
dtype='object')
sc.pl.umap(adata, color="assignments", groups=celltypes)

set(TERMINAL_STATE).intersection(np.unique(adata.obs["assignments"]))
{'BCC',
'ChC',
'Gut_glia',
'Gut_neuron',
'Melanocytes',
'Mesenchyme',
'SC',
'SatGlia',
'Sensory',
'Symp',
'enFib'}
adata.obs["all_states"] = adata.obs["assignments"].astype("str")
adata.obs["all_states"][adata.obs["all_states"] == "none"] = np.nan
adata.obs["all_states"] = adata.obs["all_states"].astype("category")
state_names = adata.obs["all_states"].cat.categories.tolist()
adata.obs["all_states"] = (
adata.obs["all_states"].astype(str).astype("category").cat.reorder_categories(["nan"] + state_names)
)
adata.uns["all_states_colors"] = ["#dedede"] + adata.uns["assignments_colors"][:12]
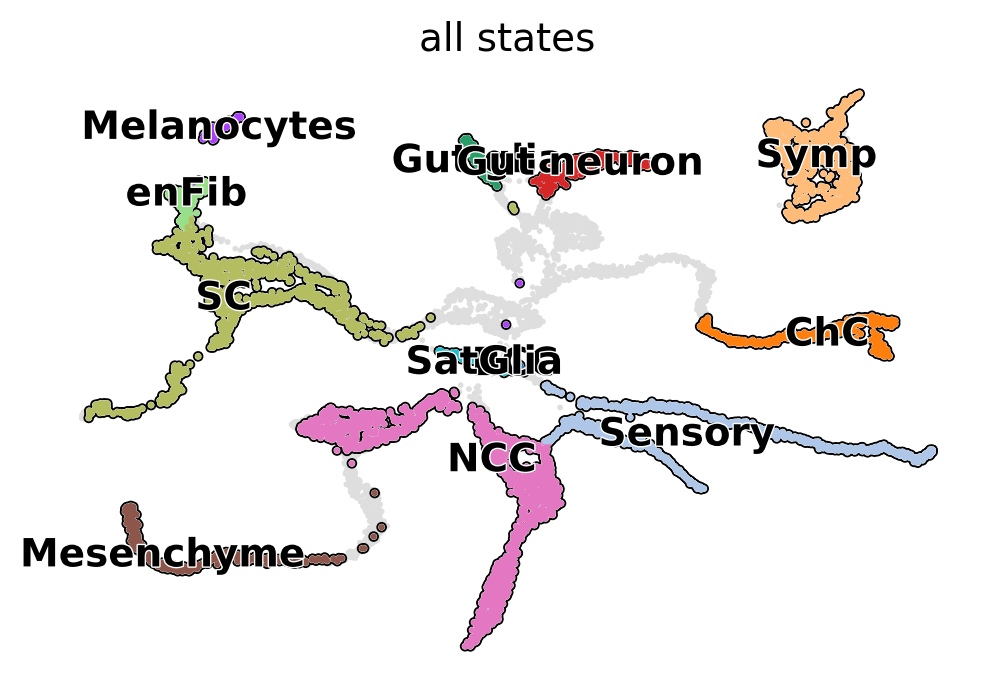
scv.pl.scatter(
adata,
basis="X_umap",
c="all_states",
add_outline=state_names,
)
Merge spliced/unspliced readout#
# adata=scv.utils.merge(adata,ldata)
# adata.obsp=None
del adata.uns["neighbors"]
adata
AnnData object with n_obs × n_vars = 8842 × 21676
obs: 'plates', 'devtime', 'location', 'n_genes_by_counts', 'total_counts', 'total_counts_ERCC', 'pct_counts_ERCC', 'doublet_scores', 'leiden', 'CytoTRACE', 'Gut_neuron', 'Sensory', 'Symp', 'enFib', 'ChC', 'Gut_glia', 'NCC', 'Mesenchyme', 'Melanocytes', 'SatGlia', 'SC', 'BCC', 'conflict', 'assignments', 'all_states'
var: 'ERCC', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'm', 'v', 'n_obs', 'res', 'lp', 'lpa', 'qv', 'highly_variable'
uns: 'devtime_colors', 'leiden', 'leiden_colors', 'leiden_sizes', 'location_colors', 'log1p', 'paga', 'umap', 'assignments_colors', 'all_states_colors'
obsm: 'X_diff', 'X_pca', 'X_umap'
layers: 'palantir_imp', 'scaled', 'GEX'
obsp: 'connectivities', 'distances'
Split into different scale level#
Location
['Cranial',
'Trunk',
'DRG',
'Limb',
'Enteric',
'Sympathoadrenal system',
'Incisor']
adatas = []
for i in range(2, 7):
adatas.append(adata[[c in Location[: (i + 1)] for c in adata.obs["location"]]].copy())
del adatas[3] # Merge last two stage
adatas
[AnnData object with n_obs × n_vars = 4012 × 21676
obs: 'plates', 'devtime', 'location', 'n_genes_by_counts', 'total_counts', 'total_counts_ERCC', 'pct_counts_ERCC', 'doublet_scores', 'leiden', 'CytoTRACE', 'Gut_neuron', 'Sensory', 'Symp', 'enFib', 'ChC', 'Gut_glia', 'NCC', 'Mesenchyme', 'Melanocytes', 'SatGlia', 'SC', 'BCC', 'conflict', 'assignments', 'all_states'
var: 'ERCC', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'm', 'v', 'n_obs', 'res', 'lp', 'lpa', 'qv', 'highly_variable'
uns: 'devtime_colors', 'leiden', 'leiden_colors', 'leiden_sizes', 'location_colors', 'log1p', 'paga', 'umap', 'assignments_colors', 'all_states_colors'
obsm: 'X_diff', 'X_pca', 'X_umap'
layers: 'palantir_imp', 'scaled', 'GEX'
obsp: 'connectivities', 'distances',
AnnData object with n_obs × n_vars = 5198 × 21676
obs: 'plates', 'devtime', 'location', 'n_genes_by_counts', 'total_counts', 'total_counts_ERCC', 'pct_counts_ERCC', 'doublet_scores', 'leiden', 'CytoTRACE', 'Gut_neuron', 'Sensory', 'Symp', 'enFib', 'ChC', 'Gut_glia', 'NCC', 'Mesenchyme', 'Melanocytes', 'SatGlia', 'SC', 'BCC', 'conflict', 'assignments', 'all_states'
var: 'ERCC', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'm', 'v', 'n_obs', 'res', 'lp', 'lpa', 'qv', 'highly_variable'
uns: 'devtime_colors', 'leiden', 'leiden_colors', 'leiden_sizes', 'location_colors', 'log1p', 'paga', 'umap', 'assignments_colors', 'all_states_colors'
obsm: 'X_diff', 'X_pca', 'X_umap'
layers: 'palantir_imp', 'scaled', 'GEX'
obsp: 'connectivities', 'distances',
AnnData object with n_obs × n_vars = 6327 × 21676
obs: 'plates', 'devtime', 'location', 'n_genes_by_counts', 'total_counts', 'total_counts_ERCC', 'pct_counts_ERCC', 'doublet_scores', 'leiden', 'CytoTRACE', 'Gut_neuron', 'Sensory', 'Symp', 'enFib', 'ChC', 'Gut_glia', 'NCC', 'Mesenchyme', 'Melanocytes', 'SatGlia', 'SC', 'BCC', 'conflict', 'assignments', 'all_states'
var: 'ERCC', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'm', 'v', 'n_obs', 'res', 'lp', 'lpa', 'qv', 'highly_variable'
uns: 'devtime_colors', 'leiden', 'leiden_colors', 'leiden_sizes', 'location_colors', 'log1p', 'paga', 'umap', 'assignments_colors', 'all_states_colors'
obsm: 'X_diff', 'X_pca', 'X_umap'
layers: 'palantir_imp', 'scaled', 'GEX'
obsp: 'connectivities', 'distances',
AnnData object with n_obs × n_vars = 8842 × 21676
obs: 'plates', 'devtime', 'location', 'n_genes_by_counts', 'total_counts', 'total_counts_ERCC', 'pct_counts_ERCC', 'doublet_scores', 'leiden', 'CytoTRACE', 'Gut_neuron', 'Sensory', 'Symp', 'enFib', 'ChC', 'Gut_glia', 'NCC', 'Mesenchyme', 'Melanocytes', 'SatGlia', 'SC', 'BCC', 'conflict', 'assignments', 'all_states'
var: 'ERCC', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells', 'm', 'v', 'n_obs', 'res', 'lp', 'lpa', 'qv', 'highly_variable'
uns: 'devtime_colors', 'leiden', 'leiden_colors', 'leiden_sizes', 'location_colors', 'log1p', 'paga', 'umap', 'assignments_colors', 'all_states_colors'
obsm: 'X_diff', 'X_pca', 'X_umap'
layers: 'palantir_imp', 'scaled', 'GEX'
obsp: 'connectivities', 'distances']
For each scale we independently build UMAP#
Following preprocessing procedure in glia fate data preprocessing code
adatas_p = []
for ad_idx in range(len(adatas)):
ad = adatas[ad_idx]
st = count_cell_types(ad.obs["assignments"], TERMINAL_STATE)
ad = ad[[i not in st["Cell Type"][st["Count"] < 30].tolist() for i in ad.obs["assignments"]]].copy()
## UMAP embedding
sc.pp.neighbors(ad, n_neighbors=80, use_rep="X_diff")
adatas_p.append(ad)
computing neighbors
2025-08-19 14:39:44.772428: E external/local_xla/xla/stream_executor/cuda/cuda_fft.cc:477] Unable to register cuFFT factory: Attempting to register factory for plugin cuFFT when one has already been registered
WARNING: All log messages before absl::InitializeLog() is called are written to STDERR
E0000 00:00:1755607185.730112 2476790 cuda_dnn.cc:8310] Unable to register cuDNN factory: Attempting to register factory for plugin cuDNN when one has already been registered
E0000 00:00:1755607186.191275 2476790 cuda_blas.cc:1418] Unable to register cuBLAS factory: Attempting to register factory for plugin cuBLAS when one has already been registered
finished (0:01:08)
computing neighbors
finished (0:00:00)
computing neighbors
finished (0:00:00)
computing neighbors
finished (0:00:18)
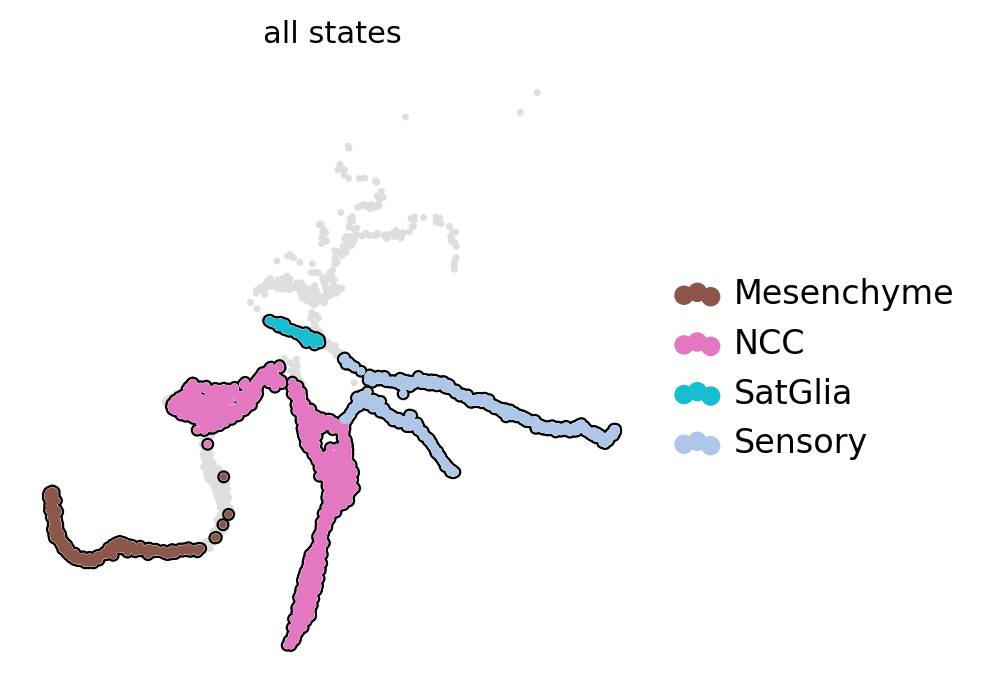
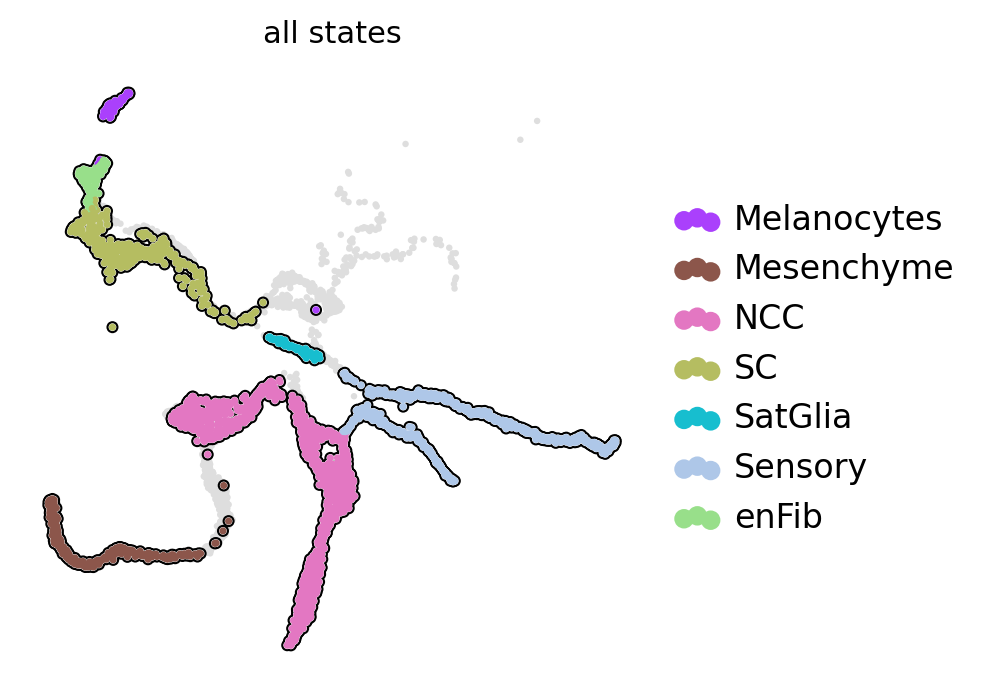
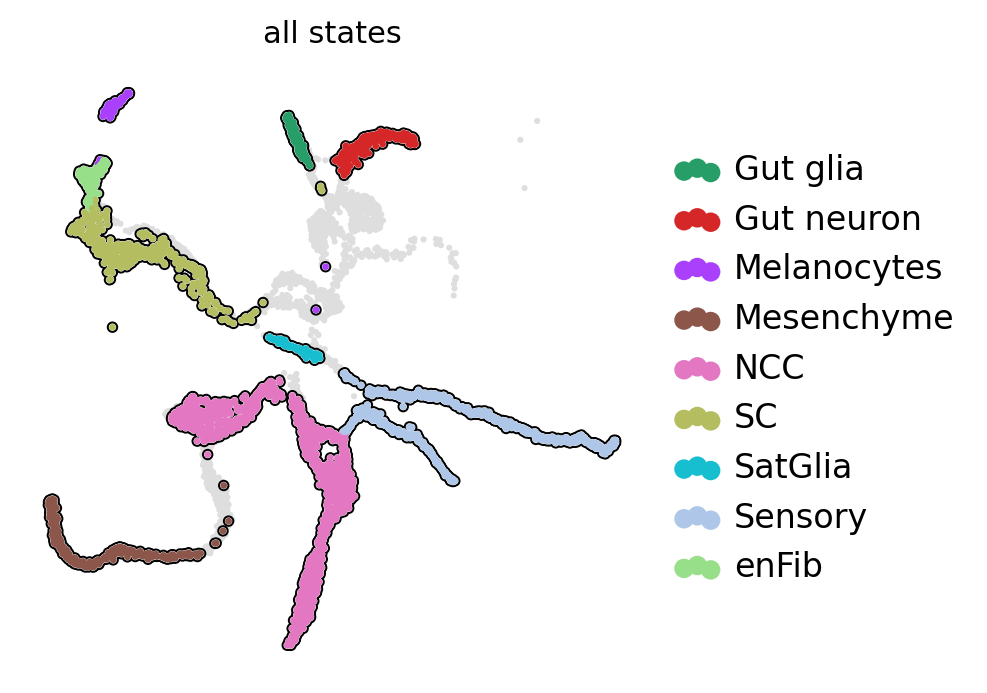
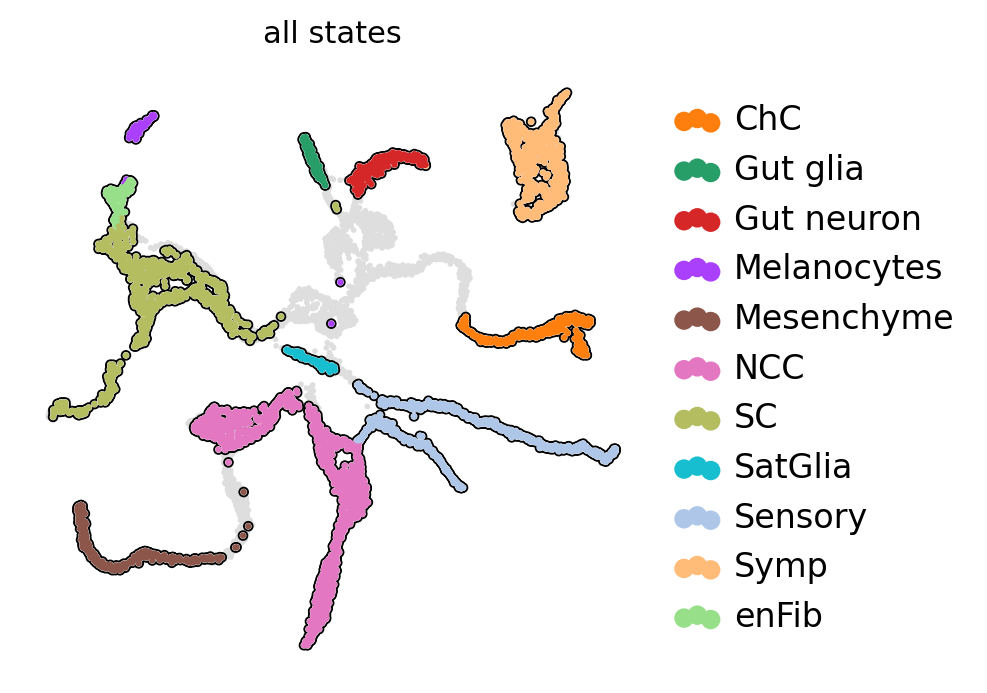
for idx in range(4):
ad = adatas_p[idx].copy()
ad.X = ad.layers["GEX"].copy()
with mplscience.style_context():
fig, ax = plt.subplots(figsize=(4, 4))
# scv.pl.scatter(adata[cells,:], c="assignments", ax=ax,frameon = False,add_outline = set(celltypes).intersection(np.unique(adata.obs["assignments"])),legend_loc = "right")
scv.pl.scatter(ad, basis="X_umap", c="all_states", add_outline=state_names, legend_loc="right", ax=ax)
if SAVE_FIGURES:
fig.savefig(
FIG_DIR / DATASET / f"scale_{idx+1}_umap.svg",
format=FIGURE_FORMAT,
transparent=True,
bbox_inches="tight",
)
if SAVE_DATA:
ad.write_h5ad(DATA_DIR / DATASET / "processed" / f"adata_stage{idx+1}_processed.h5ad")