Reannotation of pigment cells#
In our smart-seq3, although we know our pigment cells mainly express gch2 gene, we still need to annotate the actual gch2+ pigment cells to make sure our evaluated cell state align to the cell state in perturb-seq. Therefore, we could integrate the pigment cell population in perturb-seq and smart-seq3, and transfer perturb-seq label on smart-seq3 dataset.
Library imports#
import faiss
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import scanpy as sc
import scanpy.external as sce
import scvelo as scv
from rgv_tools import DATA_DIR
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/louvain/__init__.py:54: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import get_distribution, DistributionNotFound
General setting#
plt.rcParams["svg.fonttype"] = "none"
sns.reset_defaults()
sns.reset_orig()
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=14, color_map="viridis")
Constants#
DATASET = "zebrafish"
SAVE_DATA = True
if SAVE_DATA:
(DATA_DIR / DATASET / "processed").mkdir(parents=True, exist_ok=True)
Data loading#
Note: The perturb-seq datasets will be released after the paper is officially online. Users can access them via regvelo.datasets.zebrafish_perturb.
perturb_seq = sc.read_h5ad(DATA_DIR / DATASET / "raw" / "perturbseq_all.h5ad")
ss3 = sc.read_h5ad(DATA_DIR / DATASET / "processed" / "adata_preprocessed.h5ad")
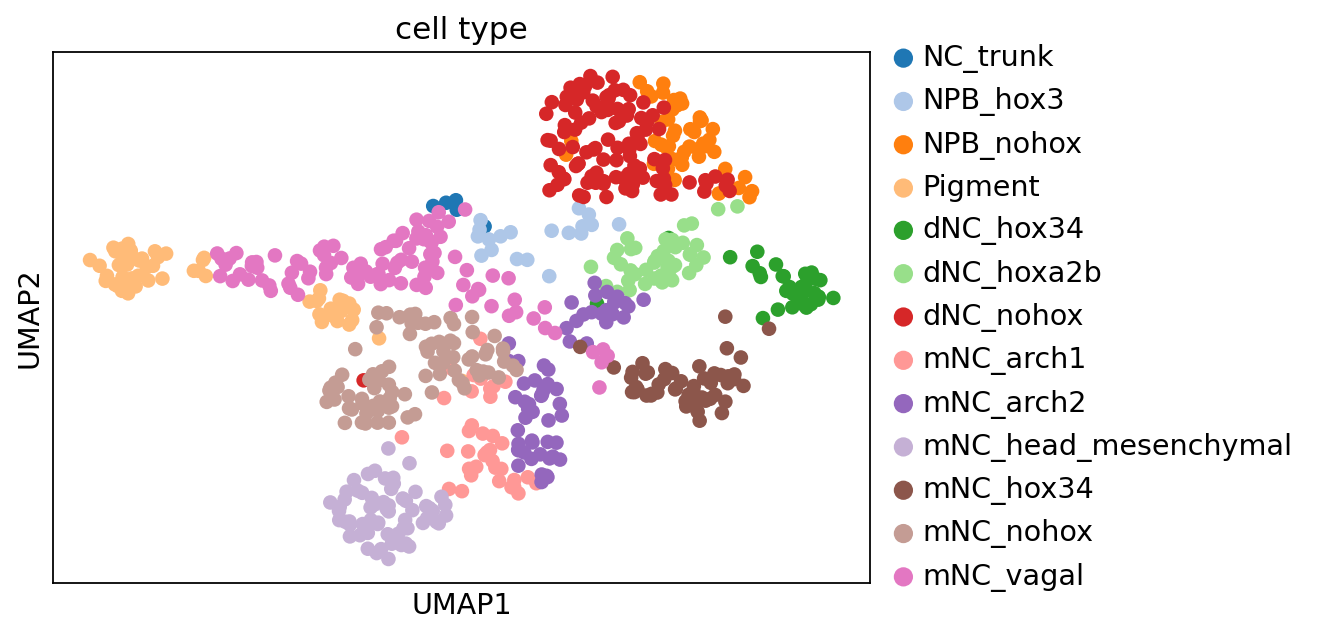
sc.pl.scatter(ss3, basis="umap", color="cell_type")
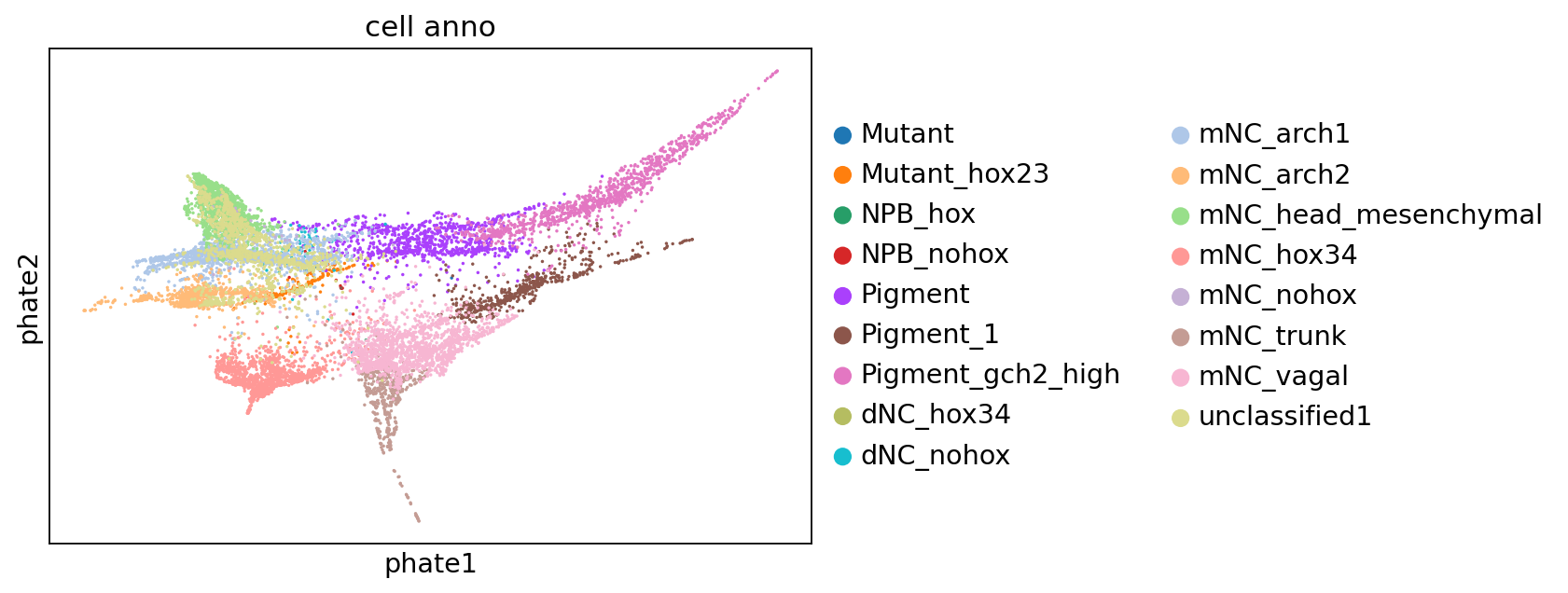
sc.pl.scatter(perturb_seq, basis="phate", color="cell_anno")
Integrate ss3 and perturb seq dataset#
ss3_p = ss3[ss3.obs["cell_type"] == "Pigment"]
ref_p = perturb_seq[perturb_seq.obs["sgRNA_group"] == "control"].copy()
ref_p = ref_p[ref_p.obs["cell_anno"].isin(["Pigment", "Pigment_gch2_high"])].copy()
sc.pp.scale(ref_p)
sc.pp.scale(ss3_p)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scanpy/preprocessing/_scale.py:318: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
gene = list(set(ss3_p.var_names).intersection(ref_p.var_names))
merged_adata = ref_p[:, gene].concatenate(ss3_p[:, gene], batch_key="batch")
sc.tl.pca(merged_adata)
sce.pp.harmony_integrate(merged_adata, "batch")
2025-08-28 16:46:47,452 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...
2025-08-28 16:46:48,060 - harmonypy - INFO - sklearn.KMeans initialization complete.
2025-08-28 16:46:48,068 - harmonypy - INFO - Iteration 1 of 10
2025-08-28 16:46:48,114 - harmonypy - INFO - Iteration 2 of 10
2025-08-28 16:46:48,135 - harmonypy - INFO - Iteration 3 of 10
2025-08-28 16:46:48,146 - harmonypy - INFO - Iteration 4 of 10
2025-08-28 16:46:48,156 - harmonypy - INFO - Iteration 5 of 10
2025-08-28 16:46:48,165 - harmonypy - INFO - Converged after 5 iterations
sc.pp.neighbors(merged_adata, n_pcs=30, use_rep="X_pca_harmony")
sc.tl.umap(merged_adata)
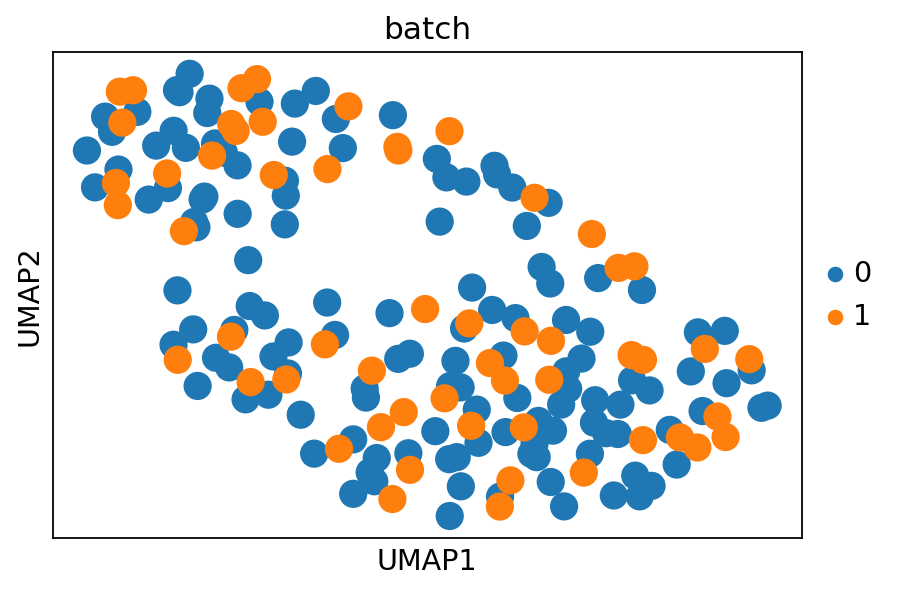
sc.pl.umap(
merged_adata,
color="batch",
# Setting a smaller point size to get prevent overlap
)
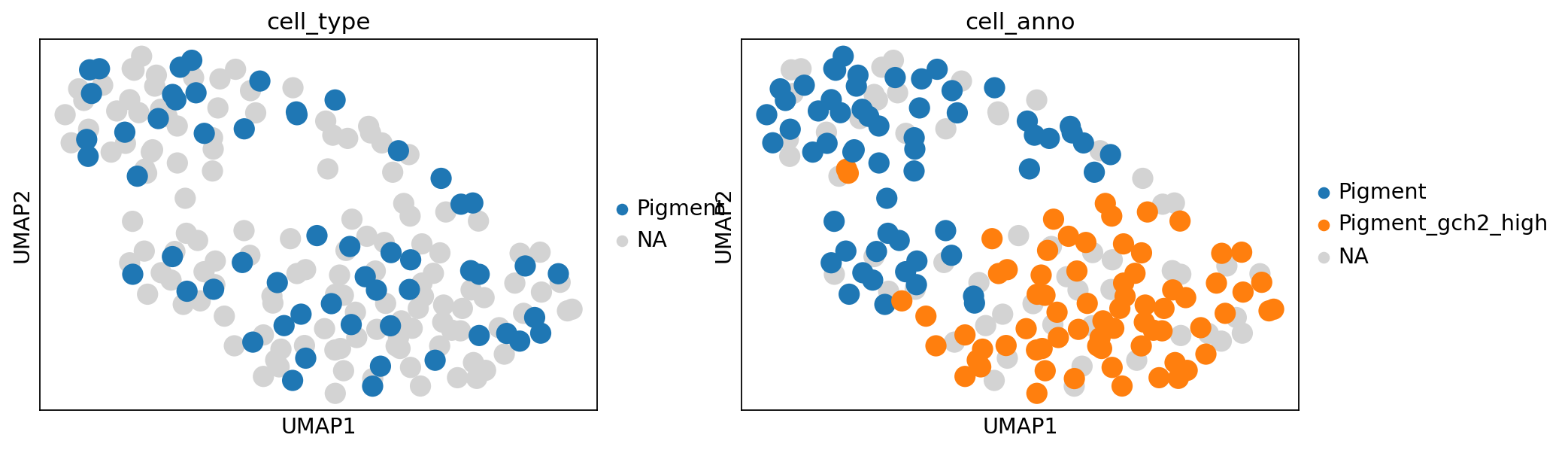
sc.pl.umap(
merged_adata,
color=["cell_type", "cell_anno"],
# Setting a smaller point size to get prevent overlap
)
X = merged_adata.obsm["X_pca_harmony"].astype("float32")
# build FAISS KNN index
n_cells = X.shape[0]
k = 10
index = faiss.IndexFlatL2(X.shape[1])
index.add(X)
distances, neighbors = index.search(X, k)
# project neighbors cell type label
sgRNA_labels = merged_adata.obs["cell_anno"].values
neighbor_labels = sgRNA_labels[neighbors]
mutant_mask = (neighbor_labels != "control").astype(int)
mutant_neighbor_counts = mutant_mask.sum(axis=1)
score = (np.array(neighbor_labels[merged_adata.obs["cell_type"].isin(["Pigment"])]) == "Pigment_gch2_high").astype(int)
score = score.sum(axis=1) / 10
annotation = pd.DataFrame(merged_adata.obs["cell_type"][merged_adata.obs["cell_type"].isin(["Pigment"])])
annotation.loc[score >= 0.7, "pigment_annotation"] = "Pigment_gch2"
Save data#
if SAVE_DATA:
annotation.to_csv(DATA_DIR / DATASET / "processed" / "annotation.csv")