Evaluate the cell fate perturbation significance on perturb-seq#
We used permutation test to evaluate the significance via shuffling the MELD likelihood
Library imports#
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import cellrank as cr
import scanpy as sc
import scvelo as scv
from rgv_tools import DATA_DIR
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_csv from `anndata` is deprecated. Import anndata.io.read_csv instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_excel from `anndata` is deprecated. Import anndata.io.read_excel instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_hdf from `anndata` is deprecated. Import anndata.io.read_hdf instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_loom from `anndata` is deprecated. Import anndata.io.read_loom instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_mtx from `anndata` is deprecated. Import anndata.io.read_mtx instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_text from `anndata` is deprecated. Import anndata.io.read_text instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/anndata/utils.py:434: FutureWarning: Importing read_umi_tools from `anndata` is deprecated. Import anndata.io.read_umi_tools instead.
warnings.warn(msg, FutureWarning)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/louvain/__init__.py:54: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import get_distribution, DistributionNotFound
General setting#
plt.rcParams["svg.fonttype"] = "none"
sns.reset_defaults()
sns.reset_orig()
scv.settings.set_figure_params("scvelo", dpi_save=400, dpi=80, transparent=True, fontsize=14, color_map="viridis")
Constants#
DATASET = "zebrafish"
SAVE_DATA = True
if SAVE_DATA:
(DATA_DIR / DATASET / "results").mkdir(parents=True, exist_ok=True)
groups = [
"fli1a_erf_erfl3",
"control",
"erf_erfl3",
"mitfa",
"rarga",
"nr2f5",
"tfec",
"rxraa",
"fli1a_elk3",
"tfec_mitfa_bhlhe40",
"elk3",
"mitfa_tfec",
"mitfa_tfec_tfeb",
"fli1a",
"ebf3a",
"elf1",
"nr2f2",
"ets1",
]
Data loading#
adata = sc.read_h5ad(DATA_DIR / DATASET / "processed" / "perturbseq_all.h5ad")
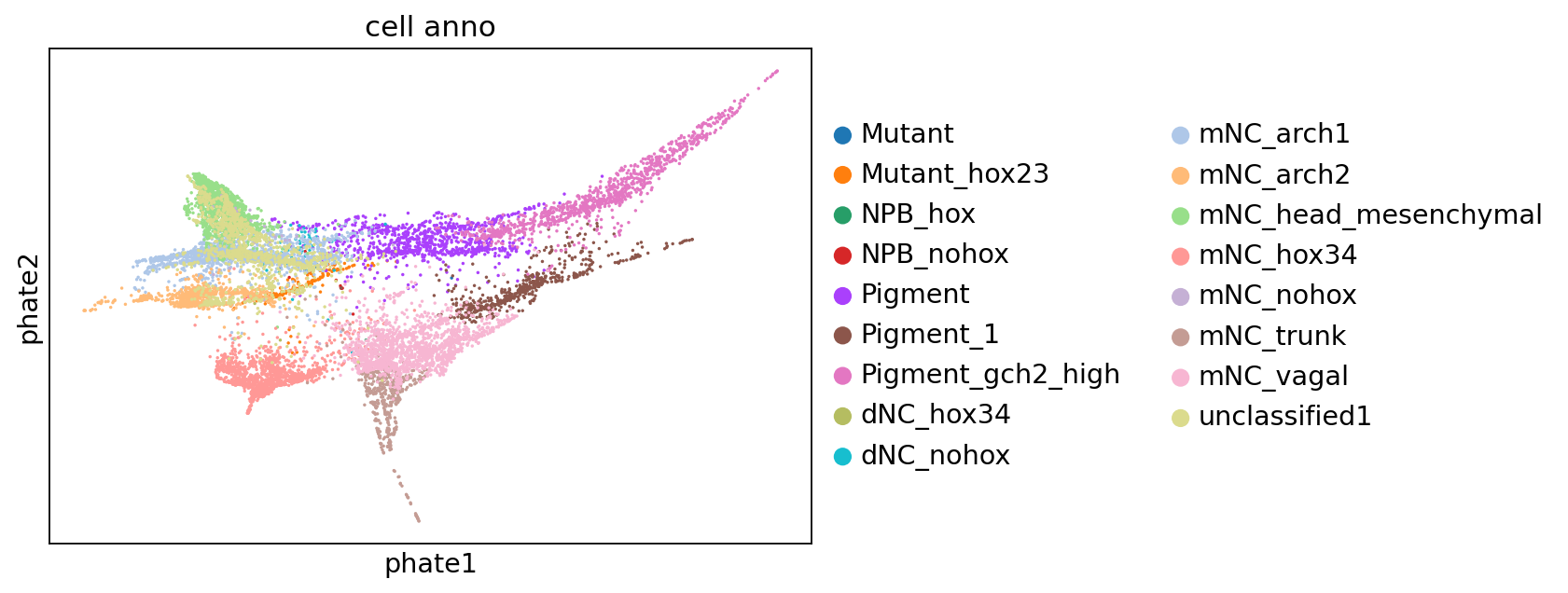
sc.pl.scatter(adata, basis="phate", color="cell_anno")
adata = adata[adata.obs["sgRNA_group"].isin(groups)].copy()
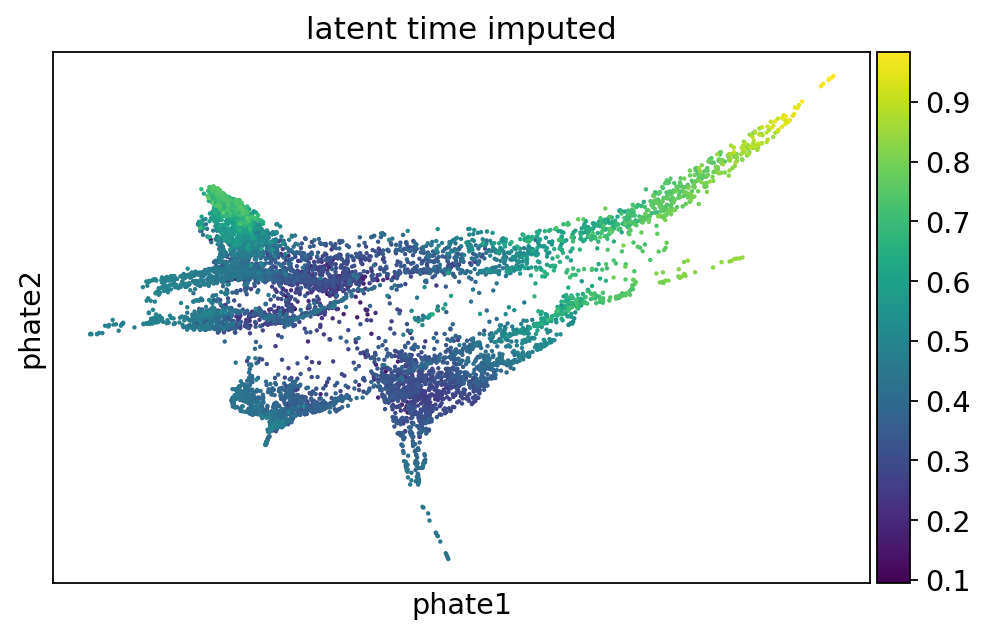
sc.pl.scatter(adata, basis="phate", color="latent_time_imputed")
sc.pp.neighbors(adata, n_pcs=30, n_neighbors=30)
n_states = 7
pk = cr.kernels.PseudotimeKernel(adata, time_key="latent_time_imputed")
pk.compute_transition_matrix()
## evaluate the fate prob on original space
estimator = cr.estimators.GPCCA(pk)
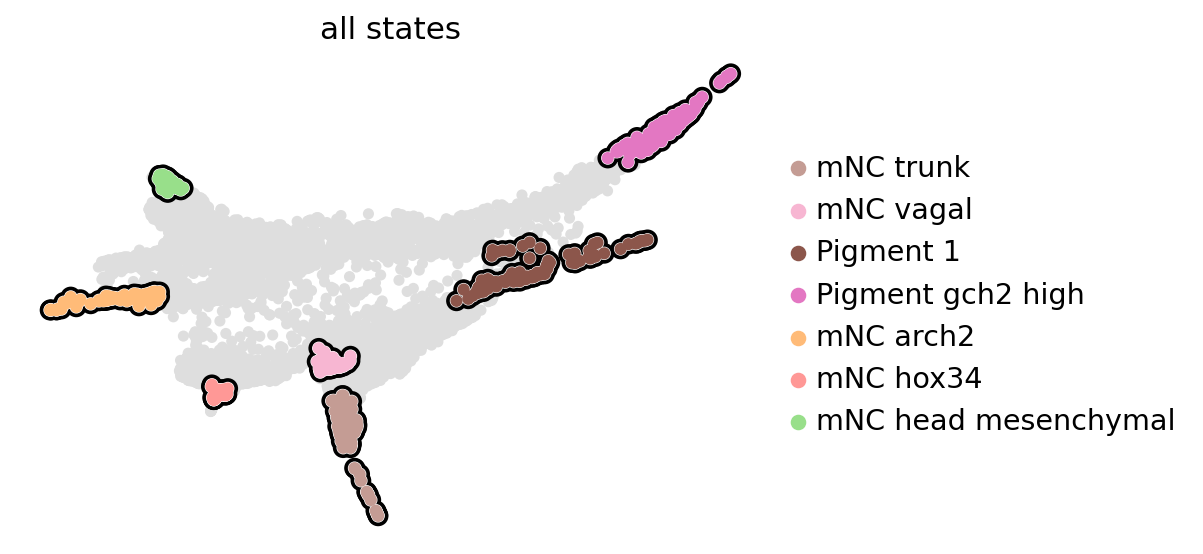
estimator.compute_macrostates(n_states=n_states, cluster_key="cell_anno", n_cells=100)
GPCCA[kernel=PseudotimeKernel[n=7676], initial_states=None, terminal_states=None]
estimator.plot_macrostates(which="all", discrete=True, legend_loc="right", s=100, basis="phate")
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/scatter.py:656: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
smp = ax.scatter(
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/scatter.py:694: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/utils.py:1396: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(x, y, s=bg_size, marker=".", c=bg_color, zorder=zord - 2, **kwargs)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/utils.py:1397: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(x, y, s=gp_size, marker=".", c=gp_color, zorder=zord - 1, **kwargs)
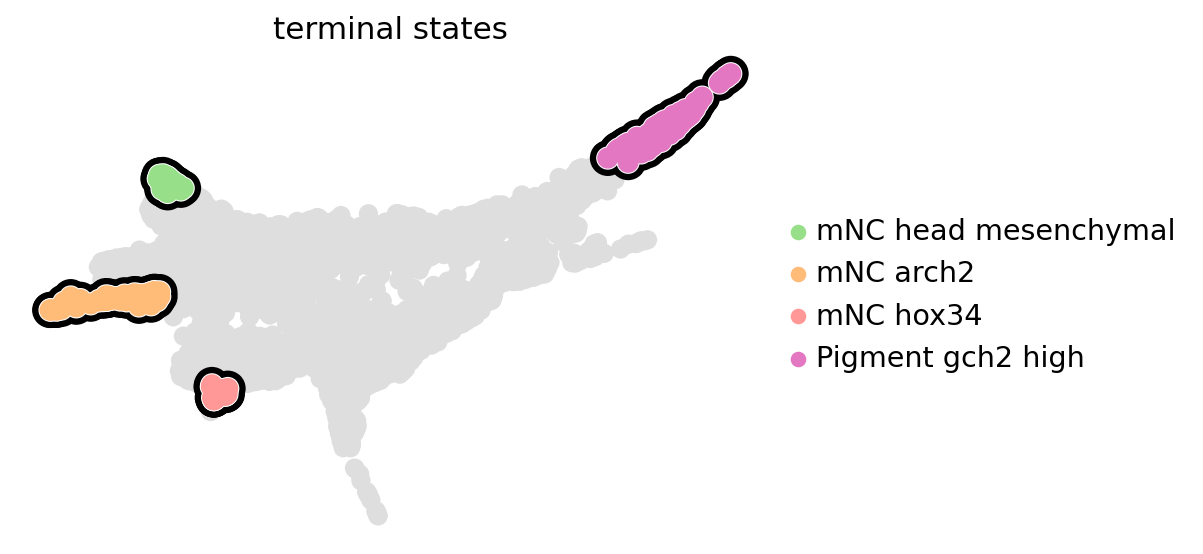
estimator.set_terminal_states(
states=["mNC_head_mesenchymal", "mNC_arch2", "mNC_hox34", "Pigment_gch2_high"], n_cells=100
)
estimator.plot_macrostates(which="terminal", legend_loc="right", size=300, basis="phate")
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/scatter.py:656: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
smp = ax.scatter(
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/scatter.py:694: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/utils.py:1396: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(x, y, s=bg_size, marker=".", c=bg_color, zorder=zord - 2, **kwargs)
/home/icb/weixu.wang/miniconda3/envs/regvelo-py310/lib/python3.10/site-packages/scvelo/plotting/utils.py:1397: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
ax.scatter(x, y, s=gp_size, marker=".", c=gp_color, zorder=zord - 1, **kwargs)
cell_states = [
"mNC_head_mesenchymal",
"mNC_arch1",
"mNC_arch2",
"mNC_hox34",
"mNC_vagal",
"mNC_trunk",
"Pigment",
"Pigment_1",
"Pigment_gch2_high",
"unclassified1",
"dNC_nohox",
"Mutant",
"Mutant_hox23",
]
score_m = []
for g in np.array(groups)[[i not in ["control"] for i in groups]]:
filename = DATA_DIR / DATASET / "raw" / f"MELD_likelihood_raw/likelihood_{g}.csv"
likelihood = pd.read_csv(filename, index_col=0)
sobject = adata[(adata.obs["sgRNA_group"] == g) | (adata.obs["sgRNA_group"] == "control")].copy()
# Assign likelihood values to the subset, aligned by column (cell) names
# sobject.obs['likelihood'] = likelihood.loc[sobject.obs.index,"likelihood"]
sobject.obs["likelihood"] = likelihood.loc[sobject.obs.index, [f"{g}A"]]
score = []
for i in ["mNC_arch2", "mNC_head_mesenchymal", "mNC_hox34", "Pigment_gch2_high"]:
# Filter by cell state
obj = sobject[sobject.obs["term_states_fwd"] == i]
# Get likelihoods for cells with latent_time_imputed >= median
lks = obj.obs.loc[:, "likelihood"]
# Append median likelihood to score vector
score.append(np.median(lks))
# Append scores to result matrix
score_m.append(score)
score_m = pd.DataFrame(
score_m,
columns=["mNC_arch2", "mNC_head_mesenchymal", "mNC_hox34", "Pigment_gch2_high"],
index=np.array(groups)[[i not in ["control"] for i in groups]],
)
MELD_score = score_m.copy()
Calculate P-value#
score_m_all = []
for _nrun in range(1000):
score_m = []
for g in np.array(groups)[[i not in ["control"] for i in groups]]:
filename = DATA_DIR / DATASET / "raw" / f"MELD_likelihood_raw/likelihood_{g}.csv"
likelihood = pd.read_csv(filename, index_col=0)
sobject = adata[(adata.obs["sgRNA_group"] == g) | (adata.obs["sgRNA_group"] == "control")].copy()
# Assign likelihood values to the subset, aligned by column (cell) names
sobject.obs["likelihood"] = likelihood.loc[sobject.obs.index, [f"{g}A"]]
sobject.obs["term_states_fwd"] = np.random.permutation(sobject.obs["term_states_fwd"].tolist()).tolist()
score = []
for i in ["mNC_arch2", "mNC_head_mesenchymal", "mNC_hox34", "Pigment_gch2_high"]:
# Filter by cell state
obj = sobject[sobject.obs["term_states_fwd"] == i]
# Get likelihoods for cells with latent_time_imputed >= median
lks = obj.obs.loc[:, "likelihood"]
# Append median likelihood to score vector
score.append(np.median(lks))
# Append scores to result matrix
score_m.append(score)
score_m_all.append(np.array(score_m))
score_perm_array = np.array(score_m_all) # shape: (1000, len(groups), 4)
# Empirical p-value: count how many times permuted scores are more extreme than observed
# Two-tailed p-value
abs_real = np.abs(MELD_score.values)
abs_perm = np.abs(score_perm_array)
# Count how many permuted scores are >= observed scores (broadcasting)
counts = np.sum(abs_perm >= abs_real[None, :, :], axis=0)
# Empirical p-value: (count + 1) / (n_permutations + 1) to avoid zero p-values
empirical_p = (counts) / (score_perm_array.shape[0])
# Convert to DataFrame with same structure
empirical_p_df = pd.DataFrame(empirical_p, index=MELD_score.index, columns=MELD_score.columns)
Save data#
if SAVE_DATA:
MELD_score.to_csv(DATA_DIR / DATASET / "results" / "MELD_median_score.csv")
empirical_p_df.to_csv(DATA_DIR / DATASET / "results" / "pval_mat.csv")